
Abstract
Background
Liquid chromatography linked to tandem mass spectrometry (LC/MS/MS) is being increasingly used for drug confirmation. At present, no official criteria exist for drug identification using this technique although the European Union (EU) criteria for compound identification have been adopted. These criteria are evaluated with respect to opiate confirmation by LC/MS/MS and problems highlighted.
Methods
Urine samples screened positive for opiates by immunoassay were subjected to confirmation by LC/MS/MS using multiple reaction monitoring (MRM) and two separate buffer systems of pH 6.8 and 8.0, respectively. The EU criteria for compound identification were applied for confirmation of morphine, 6-monoacetylmorphine (6MAM), codeine and dihydrocodeine (DHC).
Results
Using the pH 6.8 buffer, confirmation could be achieved for 84%, 94%, 96% and 95%, respectively, for samples demonstrating MRM chromatographic peaks at retention times for morphine, 6MAM, codeine and DHC. Failure to meet the EU criteria was mainly attributed to low signal-to-noise (S:N) ratios or excessively high drug concentrations. Isobaric interferences and poor chromatography were also contributing factors. The identification of morphine was considerably improved with chromatography at pH 8.0 owing to resolution of interferences. Oxycodone metabolites were a potential problem for the identification of DHC.
Conclusion
Isobaric interferences can pose a problem with drug identification using LC/MS/MS. Optimizing chromatographic conditions is important to overcome these interferences. Consideration needs to be given to investigating drug metabolites as well as parent drugs in method development.
Introduction
Confirmation of drugs and/or drug metabolites is essential in toxicological analysis, particularly where medicolegal issues apply and is also necessary to identify particular compounds following initial screening by a generic antibody, e.g. opiate immunoassay. This is highlighted in the SOFT/AAFS Forensic Laboratory Guidelines, 1 which state that a second technique for confirmation based on a different chemical principle should be used for confirmation. Gas chromatography hyphenated to mass spectrometry has been and remains to be the most popular technique for drug confirmation. However, liquid chromatography linked to tandem mass spectrometry (LC/MS/MS) is gaining increasing acceptance in drug confirmation with compound selectivity being achieved by multiple reaction monitoring (MRM) during the course of the chromatography. MRM allows many drugs to be detected within one chromatographic run by monitoring the conversion of their respective precursor ions to a product ion following fragmentation in a collision cell.
One of the key issues regarding the use of LC/MS/MS in toxicology is the criteria for compound identification. Performance criteria with respect to LC/MS/MS have been defined by the European Union (EU) in relation to pesticide residue detection 2 and by the World Anti-doping Agency (WADA) for drugs in sport. 3 Unfortunately, no specific official criteria exist with respect to LC/MS/MS used in MRM mode for illicit drug identification. Maralikova and Weinmann 4 adopted the EU criteria for their work on drugs of abuse confirmation suggesting that there should be at least four identification points per compound. These included two MRM product ions present at specified relative ion abundances, the chromatographic retention time and co-elution of a stable isotopic internal standard. Several LC/MS/MS methods for the identification of drugs of abuse in urine have been described but without complete reference to international criteria. 5–7 This may lead to the reporting of false-positives due to the co-elution of isobaric compounds. The inclusion of the MRM ion ratios has been shown to be effective in drug identification using LC/MS/MS increasing the confidence of this technique in toxicology. 4,8
We have further evaluated the EU criteria with respect to confirmation of opiates following immunoassay screening and highlight some of the potential problems that may occur.
Materials and methods
Reagents
Morphine, codeine, 6-monoacetyl morphine (6MAM), dihydrocodeine (DHC) and oxycodone and the deuterium-labelled internal standards morphine-d6, 6MAM-d3, codeine-d3 and DHC-d6 were purchased from LGC Promochem (Teddington, Middlesex, UK). Ammonium acetate was purchased from BDH (Lutterworth, UK). All other reagents were purchased from York Glassware (York, UK).
Standards and internal standards
A mixed opiate standard was prepared by spiking drug-free urine to 10,000 μg/L with morphine, 6MAM, codeine and DHC. A set of standards to enable examination of MRM ratios over a range of drug concentrations was prepared by serial dilutions to the following concentrations: 10,000, 5000, 2500, 1250, 625 and 313 μg/L. A mixed internal standard was prepared in methanol containing 3.3 mg/L 6MAM-d3, codeine-d3, DHC-d6 and 6.7 mg/L morphine-d6.
Urine specimens
All the urine specimens used were anonymized samples submitted for routine clinical drugs of abuse testing and surplus to clinical analytical requirements. For the initial part of the study, a total of 271 consecutive opiate immunoassay-positive urine samples were analysed over 10 consecutive working days. A further 89 immunoassay-positive urine samples were used for the comparison of two separate chromatographic conditions.
Sample preparation
For the immunoassay screen, urine specimens were transferred to 5 mL polystyrene test tubes and centrifuged at 3000 g for 10 min to remove sediments. For the LC/MS/MS assay, 200 μL of centrifuged urine was transferred to 96-well microtitre plates using a Tecan Miniprep75 (TecanUK, Reading, UK) followed by the addition of 25 μL internal standard mixture.
Immunoassay screen
Urine specimens were screened for the presence of opiate group of drugs using the Synchron LX20 opiate class enzyme multiplied immunoassay technique (EMIT) assay kit (Beckman-Coulter, High Wycombe, UK). Detection cut-off was 300 μg/L. All the urine specimens positive by immunoassay were subjected to qualitative opiate confirmation analysis by LC/MS/MS.
High-performance liquid chromatography
Liquid chromatography was performed using a Shimadzu system (Milton Keynes, UK) comprising a SIL-HT autosampler, 2× LC10AC pumps, a DGU-14A degasser and a CTO-10AS column oven. The column effluent was directed either to the tandem mass spectrometer or to waste via a Valco switching valve. The setup was controlled by Analyst 1.4 software (Applied Biosystems, Warrington, Cheshire, UK) which was also used for data collection and interpretation. Chromatographic separation was performed using a 150 mm × 3 mm HyPURITY C8 column (Thermo Electron Corporation, Runcorn, Cheshire, UK) fitted with a security guard column of the same packing material (Phenomenex, Macclesfield, Cheshire, UK). The column was maintained at 35°C. Two chromatographic methods were employed using buffers with a different pH. The primary chromatographic buffer system used a mobile phase A of 4 mmol/L ammonium acetate, pH 6.8 containing 5% (v/v) methanol in high-performance liquid chromatography (HPLC) grade water. Mobile phase B consisted of 1% (v/v) propan-2-ol, 0.05% (v/v) formic acid in 100% HPLC grade methanol. Gradient elution was performed at a constant flowrate of 0.8 mL/min as follows: 0% B was maintained for 2 min followed by a linear increase to 100% B over 5 min. Mobile phase B was maintained at 100% for 2 min then reduced to 0% over 0.1 min and maintained at this level for 3.4 min. The column eluate was directed to waste for the first 3 min, to the mass spectrometer for 5 min and then to waste for the remainder of the time programme. The secondary chromatographic buffer system used a mobile phase A containing 10 mmol/L ammonium acetate, pH 8.0 (pH adjusted with ammonium hydroxide) and 5% (v/v) methanol prepared in HPLC grade water. Mobile phase B was the same as the primary chromatographic buffer system. Gradient elution was performed at a constant flowrate of 0.8 mL/min as follows: 0% B was maintained for 1 min followed by a linear increase to 100% over 4 min. Mobile phase B was maintained at 100% for 1 min then reduced to 0% B over 0.1 min and maintained at this level for 2 min. Column eluate was directed to the waste for the first 2 min, to the mass spectrometer for 4 min and then to the waste for the remainder of the time programme.
Tandem mass spectrometry
The drugs were detected using an Applied Biosystems/MDS Sciex API 3000 Triple-Quad Mass Spectrometer equipped with a TurboIonSpray source (Applied Biosystems). Analytes were detected in positive ion mode using multiple reaction monitoring of the molecular ion transitions listed in Table 1. Nitrogen was used for the nebulizer, curtain and collision gases with flowrates of 10 L/min, 10 L/min and 9 L/min, respectively. The ion-spray voltage was 5 kV and temperature was 475°C. Data were collected and quantified using Analyst software version 1.4.
Multiple reaction monitoring (MRM) transitions
*Denotes primary MRM transition
Drug identification criteria
The EU criteria were used for drug confirmation following LS/MS/MS analysis. 2 Retention time criteria: the ratio of the chromatographic retention time of the analyte to that of the internal standard, i.e. the relative retention time of the analyte must correspond to that of the standard at a tolerance of ±2.5%. Relative MRM ion-intensity criteria: three MRM transitions were measured for each drug and the peak areas were integrated using Analyst 1.4. The relative MRM ion intensities were calculated with respect to the intensity of the primary MRM transition using Microsoft Excel. For each MRM transition, the relative ion-intensity in the test specimen was compared with the mean relative ion-intensity obtained from the standards within the analytical batch (n = 6). The drugs were classified as confirmed when all relative ion-intensities were within the ranges specified in Table 2.
Maximum permitted tolerances for relative multiple reaction monitoring (MRM) ion intensities according to European Union criteria for compound identification
Results
Ten consecutive batches of urine specimens submitted for routine drugs of abuse screening comprising 271 immunoassay opiate-positives were analysed by LC/MS/MS using the primary chromatographic conditions with pH 6.8 mobile phase A. The results were categorized in accordance with the EU criteria and are summarized in Table 3. Confirmation could be achieved for 84%, 94%, 96% and 95%, respectively, of samples demonstrating chromatographic peaks at the retention times for morphine, 6MAM, codeine and DHC. Overall, failure to meet the EU criteria with regard to relative MRM ion-intensities could be mainly attributed to low signal-to-noise ratio (S:N <10) for one or more MRM transition for all opiates. Morphine showed a particular problem with regard to meeting the criteria in samples where concentrations of drug were greater than the top standard (>10,000 μg/L). Such samples could cause detector saturation leading to distortion of relative ion-intensities and column overload resulting in a slight shift in retention time. This was demonstrated by dilution of these samples within the limits of the standard curve, which corrected both the retention times and the relative MRM ion-intensities. Some chromatographic peaks in MRM mode occurring at the retention time of morphine showed poor peak shape suggesting possible isobaric interferences (Figure 1a). In contrast, peak resolution for 6MAM, codeine and DHC was excellent overall with only one sample in each case showing either a possible isobaric interference or poor chromatography. In the case of DHC, isobaric interference occurred in a patient who was later discovered to be taking oxycodone; this resulted in further experimental work whose results are discussed later.
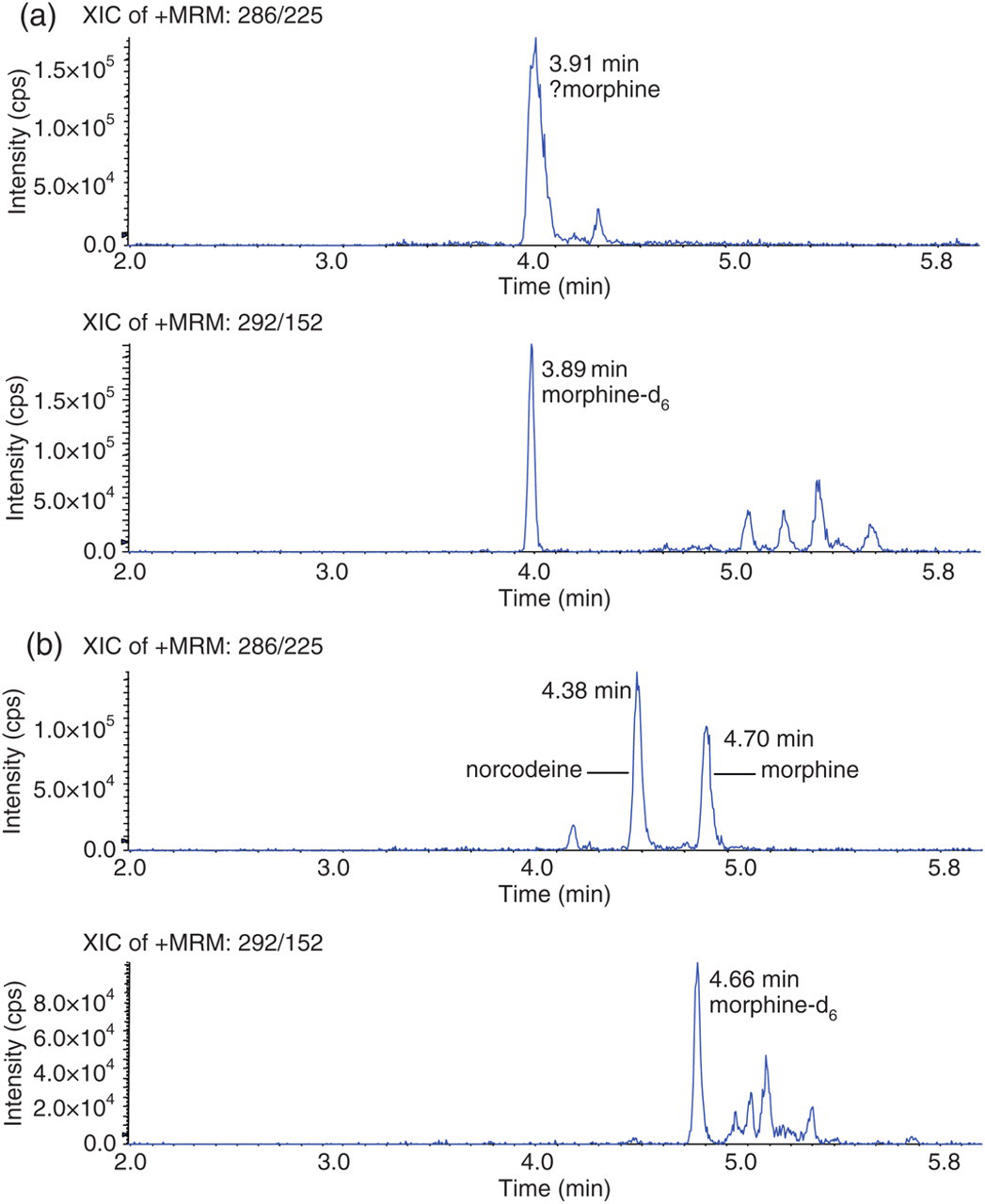
Effect of aqueous mobile phase pH on chromatography at morphine multiple reaction monitoring (MRM) transitions in a typical patient urine specimen. An opiate immunoassay-positive urine specimen was spiked with morphine-d6 to 800 μg/L and analysed under pH 6.8 (a) and pH 8.0 (b) chromatographic conditions
Summary of liquid chromatography linked to tandem mass spectrometry (LC/MS/MS) results for opiate immunoassay-positive samples with respect to European Union (EU) criteria
MRM, multiple reaction monitoring; 6MAM, 6-monoacetylmorphine; DHC, dihydrocodeine
*Possible isobaric interference or poor chromatography
The problems in relation to morphine identification with respect to the EU criteria were improved by changing the aqueous mobile phase to the secondary chromatographic buffer system (pH 8.0). The comparative data from 89 opiate-positive specimens analysed at both pH 6.8 and pH 8.0 are shown in Table 4. In this study, 97% of samples with MRM chromatographic peaks at the retention time of morphine met the EU criteria with respect to morphine using the pH 8.0 buffer versus 59% of such samples using the pH 6.8 buffer system. This improvement appeared to be due to better chromatographic resolution of morphine mainly from the isobaric compound norcodeine (m/z 268). This was apparent from the examination of both a spiked standard solution (Figure 2) and a patient sample demonstrating poor peak shape at pH 6.8 re-analysed at pH 8.0 (Figure 1). In both cases, norcodeine was clearly resolved from morphine at pH 8.0. In relation to the other opiates, altering the chromatographic conditions produced very little change with respect to the EU criteria for 6MAM. There was some improvement in codeine identification but DHC was more disappointing at pH 8.0. The problem with DHC was that patient samples were of very high concentration (>10,000 μg/L) and easily produced detector saturation. This together with variation in signal intensity from batch to batch could result in unreliability of confirmation of drugs at very high concentrations using the relative MRM ion intensities. Overall increased signal sensitivity was noted at pH 8.0 due to improved chromatographic retention of all opiates (Figure 2). Hence, drugs were eluting at higher concentrations of methanol increasing ionization in the source which would enhance signal intensity.

Chromatography of the opiates morphine, codeine, 6-monoacetylmorphine (6-MAM), dihydrocodeine (DHC) and their deuterated analogues under pH 6.8 and pH 8.0 conditions. Drug-free urine was spiked with morphine, norcodeine, codeine, 6MAM and DHC to 500 μg/L, morphine-d6 to 800 μg/L and codeine-d3, 6MAM-d3 and DHC-d6 to 400 μg/L and analysed under pH 6.8 (a) and pH 8.0 (b) chromatographic conditions
Summary of liquid chromatography linked to tandem mass spectrometry (LC/MS/MS) results for 89 opiate immunoassay-positive samples analysed under two different chromatographic conditions
Results viewed with respect to European Union criteria. MRM, multiple reaction monitoring; MAM, monoacetylmorphine; DHC, dihydrocodeine
The sample from a patient taking oxycodone which resulted in an isobaric interference at the DHC MRMs was further investigated. The presence of oxycodone in the sample was confirmed by LC/MS/MS at three MRM transitions (Figure 3a and b and Table 5). The interfering peak which was eluted with a retention time close to the deuterated DHC internal standard was present in all three DHC MRM transitions (Figure 3c and d) The EU criteria were applied to this peak as shown in Table 6. The relative ion intensities and retention time tolerances were outside the acceptable limits thus confirming that it was not DHC. Examination of the metabolic pathway of oxycodone revealed that this interfering molecule is likely to be either oxymorphone or noroxycodone (Figure 4). Both metabolites have a molecular mass of 301.34 which is within one actual mass unit of DHC (301.38). The similarity in molecular structure of these opiate drugs is likely to result in the production of precursor and product ions with very close m/z. The majority of oxymorphone in urine is conjugated to glucuronic acid and as this assay does not involve any hydrolysis of the urine samples, free oxymorphone should not be detected in the urine of subjects taking oxycodone (9). In practice, free oxymorphone could not be detected in this urine sample by LC/MS/MS. A standard solution of oxymorphone (500 μg/L) was found to elute at a retention time of 4.56 min which was faster than the peak mimicking DHC at 5.08 min. As noroxycodone is the major metabolite of oxycodone and very similar in structure to DHC and is excreted both as free and conjugated metabolites in urine, we assume that it accounts for the peak running close to the deuterated DHC standard.
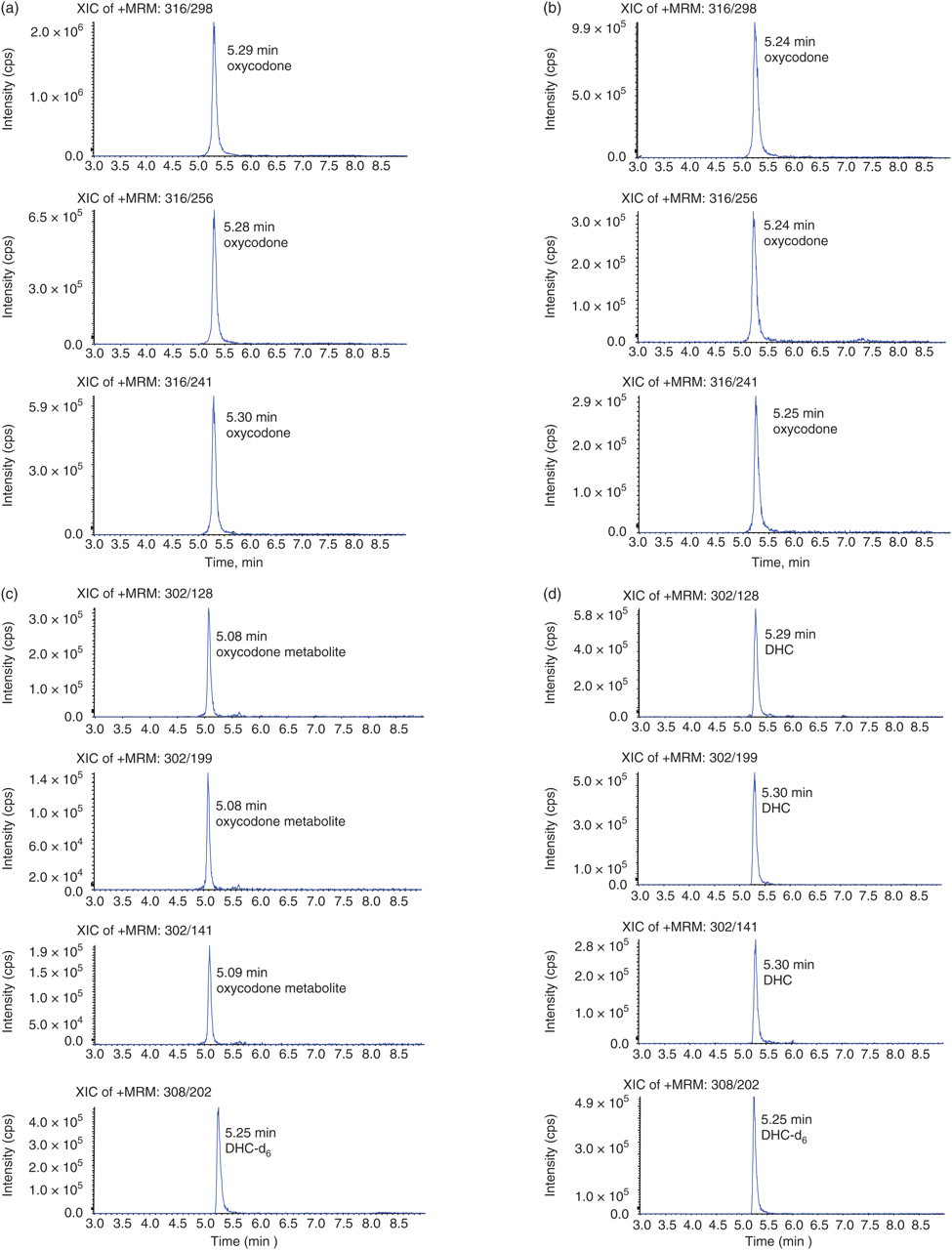
Chromatography of oxycodone, oxycodone metabolite, dihydrocodeine (DHC) and DHC-d6. Urine from a subject prescribed with oxycodone was spiked with DHC-d6 to 400 μg/L and run against DHC (500 μg/L) and oxycodone (500 μg/L) spiked standards. (a) Patient specimen at oxycodone multiple reaction monitoring (MRM) transitions m/z 316>298, 316>256 and 316>241. (b) Oxycodone-spiked blank urine at multiple reaction monitoring (MRM) transitions described in (a). (c) Patient specimen at DHC MRM transitions m/z 302>128, 302>199 and 302>141 and DHC-d6 MRM transition m/z 308>202. (d) DHC-spiked blank urine at MRM transitions described in (c)
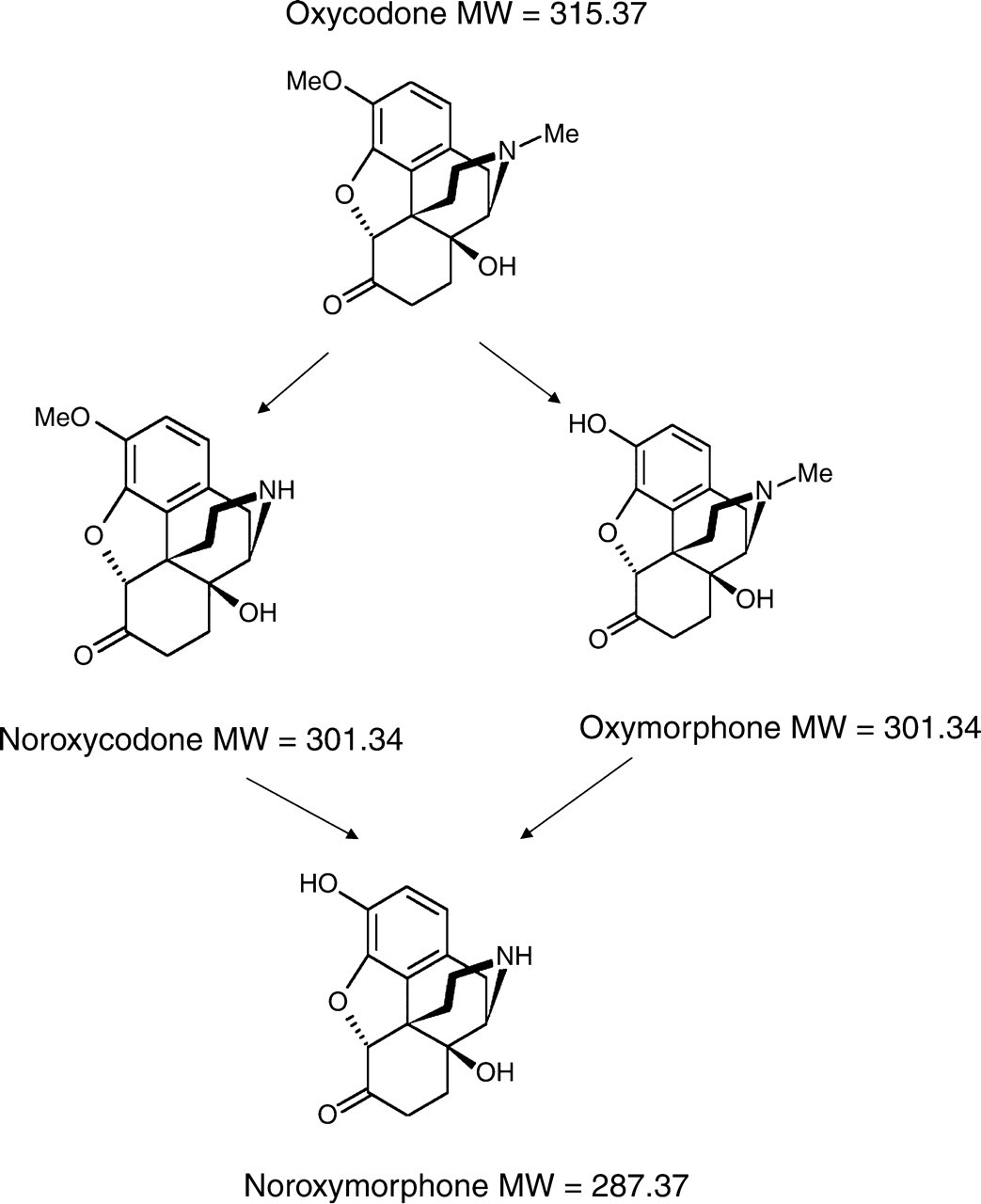
Major metabolic pathways of oxycodone. Adapted from Lalovic et al. 9
Relative multiple reaction monitoring (MRM) ion-intensities for oxycodone in patient sample and oxycodone reference standard
*Mean relative MRM ion intensity for oxycodone standard (n = 3)±25%
Relative multiple reaction monitoring (MRM) ion intensities for unknown peak occurring at dihydrocodeine (DHC) retention time compared with DHC standard
*Mean relative MRM ion intensity for DHC standard (n = 3)±20%
†Mean relative MRM ion intensity for DHC standard (n = 3)±25%
Discussion
This work clearly highlights the influence of compound identification criteria and chromatographic conditions on opiate confirmation using LC/MS/MS. We have shown that good chromatographic methods are necessary to prevent false-negative reporting owing to co-eluting isobaric compounds and that the use of retention time and relative ion intensity criteria are essential to identify unexpected interfering compounds. The importance of using more than one MRM transition to increase the specificity of drug identification was demonstrated by the work of Nordgren et al. 10 They were able to screen for 23 analytes covering the phenylethylamines and benzylpiperazine and non-benzylpiperazine hypnotic drugs using one MRM transition. One-third of samples were found to be false-positives using a confirmatory MRM transition. These authors also suggested that drug specificity could be increased by the consideration of improved chromatography, better sample preparation and consideration of drug cut-off concentrations. However, even taking these factors into account, Coles et al. 7 still found problems with isobaric interferences which could not be resolved with changes in chromatographic conditions or the use of solid-phase extraction. In this latter study, oxymorphone was found to give a false elevation in morphine and norcodeine falsely elevated hydromorphone levels. It is therefore very apparent that the structural similarities of the opiates pose a problem for drug identification using LC/MS/MS as also indicated in our study. Our work and that of others has demonstrated that the application of international compound identification criteria adds a further level of confidence for the identification of drugs of abuse. Maralikova and Weinmann 4 and Concheiro et al. 8 presented validated methods for a wide range of illicit drugs that satisfied the EU criteria. Coles et al. 7 suggested that the presence of co-eluting isobaric compounds could be detected by virtue of their ion-intensity ratios. From our finding we would suggest that drug metabolites as well as the parent compounds be taken into consideration with regard to method development. In addition to causing analytical interference, such metabolites can also be beneficial and be used as part of the drug confirmation protocol as previously shown with the confirmation of tramadol by LC/MS/MS. 11
We have shown that careful optimization of chromatographic conditions can eliminate interference from isobaric compounds. A further solution would be to increase the mass resolution of the MS/MS instrument to reduce interference from compounds of similar molecular mass. For this study unit mass resolution was used to maintain maximum sensitivity, but on more sensitive instruments, a mass resolution of down to 0.2 atomic mass units (amu) can be achieved. This may be helpful in discriminating some isobaric interferences but may not eliminate all, e.g. DHC and noroxycodone (mass difference 0.04 amu) and morphine and norcodeine (identical molecular mass).
One of the limitations of using confirmatory MRM transitions is the requirement for adequate sensitivity in terms of signal:noise for all transitions used in confirmation. This requirement forms part of the WADA criteria which stipulate that the least abundant product ion must be present with a signal:noise of at least 3. 3 In practice, MRM transition product ions vary widely in their relative abundance and the most specific are not necessarily the most sensitive. When selecting the best MRM transitions for use in quantification and confirmation, this needs to be taken into account and a compromise made to obtain both optimal specificity and adequate sensitivity. It is also essential that the relative MRM ion-intensities of compounds found in the test samples are compared with a matrix-matched standard analysed within the same batch. Concheiro et al. 8 investigated the precision of calculated relative ion-intensities on 12 drugs of abuse and found a good within-batch reproducibility (CV% <10), whereas batch-to-batch reproducibility was highly variable (CV% range 4–39%).
There have been several published studies demonstrating the advantages of LC/MS/MS over GC-MS techniques for use in illicit drug testing. 4,7 GC-MS has been and still remains to be the gold standard for drug identification due to superior chromatographic resolution and full scan fragmentation with library matching of every compound detected. The introduction of LS/MS/MS with ion-trapping capability may produce results analogous to GC/MS with full scan fragmentation patterns of selected compounds. This is rapidly becoming the technology of choice for clinical laboratories for compound identification as it couples the convenience of liquid chromatography with a high specificity of compound identification.
For the routine drugs of abuse screening service, requiring high-throughput analysis for a limited range of drugs, this level of compound identification is generally not necessary. This is where the measurement of two or three MRM transitions per compound and application of acceptance criteria is most advantageous as it enables rapid, sensitive, quantitative and confident data collection. The manufacturers of LC/MS/MS instruments are taking this on board by introducing analysis software which automates the application of drug identification criteria and improves confidence in compound identification to internationally accepted standards without the need for complex data acquisition and interpretation. The introduction of such tools should improve compound identification but the analyst still needs to be aware of potential interferences and have the appropriate chromatographic skills for resolving any problems. It is also advisable that the criteria used for compound identification using LC/MS/MS are clearly communicated when reporting results.