
Abstract
Abstract
Background
Plasma amyloid β-peptide (Aβ) can compromise the blood-brain barrier, contributing to cerebrovascular alterations and amyloid angiopathy in Alzheimer's disease (AD). The objectives of this study were to investigate the distribution of lipoprotein-bound plasma-Aβ isoforms.
Methods
This involved a case-control study of subjects with AD or amnestic mild cognitive impairment (MCI) versus controls. Lipoprotein Aβ distribution was determined in fasted plasma. For assessment of chylomicron homeostasis in the postabsorptive state, subjects were bled 4 h after a low-fat meal. The main outcome measures were plasma lipoprotein Aβ isoform distribution and lipid homeostasis.
Results
We found the majority of plasma Aβ to be associated with triglyceride-rich lipoproteins (TRLs) encompassing chylomicrons, VLDL and IDL. For all lipoprotein groups, Aβ1–40 was the predominant isoform, accounting for approximately 50% of the total. Thereafter, equivalent amounts of the isoforms 1–42, 2–40, 1–38, 1–37 and 1–39 were found. Aβ1–37, Aβ1–38 and Aβ2–40 isoforms were significantly enriched within the TRL fraction of AD/MCI subjects and similar trends were observed for isoforms Aβ1–39, Aβ1–40 and Aβ1–42. Lipoprotein-Aβ was inversely associated with plasma total- and LDL cholesterol. AD/MCI subjects were not dyslipidaemic, however, there was evidence of accumulation of chylomicrons in the postabsorptive state.
Conclusions
Our data show that Aβ was found to be associated with plasma lipoproteins, especially those enriched with triglyceride. We find that Aβ may be increased in normolipidaemic AD subjects, commensurate with possible disturbances in postprandial lipoprotein homeostasis.
Introduction
In Alzheimer's disease (AD), pathological alterations of the cerebral vasculature accompany amyloid angiopathy. 1,2 Plasma proteins, including large lipoprotein complexes, have been detected in the parenchyma of AD brains and coupled with evidence of perivascular astrocytes and activated microglia 3 , these observations are concordant with the breakdown of the blood-brain barrier (BBB). An exaggerated concentration of plasma beta-amyloid (Aβ) may contribute to the diminution of endothelial integrity and exacerbate cerebral Aβ load. Consistent with this possibility, intravascular infusions of Aβ led to significantly increased BBB porosity and cerebral inflammatory sequelae. 4–6
In biological fluids, Aβ is normally associated with lipoproteins, but a comparison of its distribution in subjects with AD is not known. In vitro studies showed that radiolabelled Aβ distributed with a range of plasma proteins including albumin. 7–10 Ghiso 11 suggested that apolipoprotein (apo) J might be a primary vehicle of Aβ transport, based on its substantial affinity for the protein in vitro. However, most of the interest has been focused on the association of Aβ with apo E-rich lipoproteins, because of strong evidence which links the increased incidence of AD in subjects with selected isoforms of this apolipoprotein. 12–16 The lipidation status of Aβ transporters may also influence burden in mice with AD, primarily through modulation of Aβ deposition or clearance. The adenosine triphosphate-binding cassette superfamily of transporters are pivotal in modulating apo E lipidation as well as intracellular sterol homeostasis, and presently there is significant focus on how this may influence AD pathogenesis. 17–18
Lipidated apo E is normally associated with posthydrolyzed remnants of the triglyceride-rich lipoproteins (TRLs), VLDL and chylomicrons, which transport endogenous and exogenous lipids, respectively. 19 The association of Aβ with apo E-rich lipoproteins raises the possibility that in a physiological context Aβ simply serves as a regulatory metabolic component of lipoproteins. 20 However, there may be pathological sequelae if its concentration or distribution becomes significantly altered. Epidemiological studies are consistent with this notion. Lipogenic diets rich in saturated fat compromise vascular integrity and are positively related to AD. 21,22
There is accumulating evidence that significant plasma transport of Aβ is linked to the genesis and secretion of TRLs including chylomicrons (lipoproteins made by the small intestine in response to dietary fats) and hepatically derived VLDL. First, in clinical studies following ingestion of a fatty meal, there is a transient increase in the plasma concentration of the Aβ precursor protein. 23 We reported that Aβ is strongly expressed in the absorptive epithelial cells of the small intestine, the site of chylomicron assembly, and its abundance is increased in response to high-fat feeding regimens. 24 In other studies, that radioiodinated Aβ tightly binds with the core lipid components of chylomicrons. When injected intravenously, these particles were taken up by fat-rich tissues including adipose tissue, the liver, bone marrow, in addition to small, but nonetheless significant concentrations of cerebral uptake. 25
Collectively, the integrity of the BBB may be compromised as a consequence of exaggerated exposure to peripheral Aβ; however, its distribution among plasma lipoproteins has not been reported. In this study, we determined the lipoprotein-bound isoforms of Aβ in subjects with AD or with mild cognitive impairment (MCI) and in age-matched controls.
Materials and methods
Subjects
Accredited institutional Human Ethics Committees of participating centres where subject recruitment occurred approved all protocols described. Thirty subjects were recruited in total – 10 with AD, 10 with MCI and 10 control subjects. Subjects with AD and MCI were recruited from an outpatient-based Memory Clinic. Subjects had a complete clinical assessment including physical and neuropsychological aspects. Subjects were diagnosed as having AD on ICD 10 criteria. Subjects were diagnosed as having amnestic MCI by described criteria 26 that included a memory complaint noticed by the subject or their family members, memory impairment 1.5 SDs below normal for age, but otherwise no other cognitive deficits and no functional impairment. Informed consent was obtained from all subjects; however for subjects in the AD group a care-giver or guardian was required in addition to give consent. Subjects taking lipid-lowering medications or other agents that may influence lipid metabolism such as hormone-replacement therapy or steroids were excluded from the study. Additional exclusion criteria included: genetic dyslipidaemia, diabetes mellitus, endocrine disorders, major systemic illness, gastrointestinal or liver abnormalities, subjects who had smoked within the last 10 years or those who had a cardiovascular event within the last six months, unusually high concentrations of physical activity within six months prior to study, sedentary or physically incapacitated.
Anthropometric measurements
Body weight and height were measured on the day of blood collection with the subjects in light clothing using standardized procedures and using a single trained observer.
Blood collection and tests performed
Subjects were required to fast overnight for 12 h before collection of blood samples in the morning. Following blood collection, samples were kept on ice prior to immediate analysis by PathWest Pathology, Royal Perth Hospital. For blood drawn from subjects in the postabsorptive state (non-fasting), the subjects were provided with a standardized breakfast containing 20 g of dairy fat and blood was drawn 4 h thereafter. Plasma samples were stored at −80°C.
Fasting plasma lipids were measured in an accredited laboratory through routine enzymatic colorimetric procedures (Roche Diagnostics, Mannheim, Germany). The coefficient of variation (CV) for cholesterol and triglyceride assays was <4%. HDL cholesterol was determined following the precipitation of apo B-containing lipoproteins and LDL cholesterol was determined indirectly using the Friedewald equation.
Serum insulin was quantified using an Immulite 2000 insulin assay (Diagnostic Products Corporation, Los Angeles, CA, USA), which is based on an immunometric ‘sandwich’ assay procedure (CV <5%); and glucose concentrations by Gluco-quant (Roche Diagnostics, Mannheim, Germany) with a CV <4%. Total apo B was measured in serum samples by immunonephelometry (Dade Behring Inc., Marburg, Germany) with a CV of <5%.
Apolipoprotein B48 was quantified directly from plasma using a Western blotting/enhanced chemiluminescence (ECL) procedure as previously described, CV <4%. 27 Briefly, following electrophoresis of plasma in Tris-acetate (3–8%) gels were precast according to manufacturers' instructions (Invitrogen, Mount Waverly, Victoria, Australia). The separated proteins were electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, North Ryde, Australia). Detection of bound apo B was achieved following incubation with an antihuman apo B antibody (DAKO A/S, Glostrup, Denmark) coupled to ECL reagent (Amersham, Little Chalfont, UK).
Lipoprotein isolation
Lipoprotein fractions were isolated from unfrozen plasma by serial ultracentrifugation, essentially as described in Havel et al. 28 The TRL, which included chylomicrons and their remnants in addition to hepatically derived VLDL and IDL, were isolated at density ≤1.019 g/mL. LDL and HDL were defined as the density fractions (1.020–1.063 g/mL and 1.064–1.21 g/mL), respectively. For isolation of TRL, ultracentrifugation was done at 229,190 g (R max) (Beckman 70.1 Ti fixed angle rotor at 50,000 rpm) at 18°C for 24 h. Following density adjustment of the infranatant, LDL was isolated at 229,190 g at 18°C for 24 h. For the separation of HDL, ultracentrifugation was performed for 39 h at 112,303 g (35,000 rpm Beckman 70.1 Ti Rotor). Lipoprotein fractions were dialyzed extensively against physiological saline containing 1 mmol/L EDTA (pH 7.4). Thereafter, samples were purged with ultrapure argon before being sealed and stored at −80°C.
Separation and quantitation of beta-amyloid
For immunoprecipitation, magnetic sheep antimouse IgG Dynabeads M-280 (Dynal, Hamburg, Germany) were incubated overnight at +4°C with monoclonal Aβ amino terminal-selective antibody, 1E8 (Schering AG, Berlin, Germany) according to the manufacturer's protocol. Eight hundred microlitres of frozen/thawed plasma lipoprotein fractions were added to 200 μL of five-fold concentrated radioimmunoprecipitation assay detergent (RIPA) buffer, 30 and 25 μL of the preactivated magnetic Dynabeads (1 μL monoclonal antibody (mAb) 1E8/1.68 × 107 beads). Samples were then incubated overnight at +4°C under rotation, following washing of the beads four times with phosphate-buffered saline/0.1% of bovine serum albumin and once with 10 mmol/L Tris–HCl (pH 7.4). For Aβ-sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE)/immunoblot, bound Aβ peptides were eluted by heating the samples to 95°C for 5 min with 25 μL of sample buffer.
For the separation of Aβ peptides, the urea version of the Bicine-/bis–Tris/Tris/sulphate SDS–PAGE was used as previously described. 29,30 Briefly, immunoprecipitated plasma lipoprotein fractions (5 μL of immunoprecipitate) in a sample buffer were applied to gel slots. Gels were run at room temperature for 1 h at a constant current of 24 mA/gel using the MiniProtean II electrophoresis unit (Bio-Rad, Munich, Germany). Next, semi-dry Western blotting was performed using PVDF membranes. Immunostaining was performed by the overnight incubation of the membranes with monoclonal amino terminal-selective antibody, 1E8 (Schering AG) at +4°C. After the washing step, the membranes were incubated for 1 h at room temperature with an antimouse-biotinylated antibody (Vector Laboratories, Burlingame, CA, USA), washed and horseradish peroxidase-coupled streptavidin (Amersham Pharmacia, Uppsala, Sweden) was added for 1 h. After the final washing step, the chemiluminescence was visualized with ECLPlus solution (Amersham Pharmacia) according to the manufacturer's protocol, using the charge coupled device (CCD) camera system (FluorSMax MultiImager, Bio-Rad).
Results
The baseline anthropometric and haemodynamic characteristics for subjects with AD, MCI and for control subjects are given in Table 1. The three groups were matched well for age, weight, height and body mass index (BMI) with no significant difference in these parameters between groups. Subjects with AD and MCI were normolipidaemic with similar concentrations of total, LDL-cholesterol and HDL-cholesterol and of plasma triglyceride. Total apo B, indicative of the sum of all non-HDL lipoproteins, is also found to be similar for the three groups of subjects.
Anthropometry and selected plasma clinical biochemistry for subjects with Alzheimer's disease (AD) (n = 10), mild cognitive impairment (n = 10) and age-matched controls (n = 10)
Subjects were diagnosed as having AD on ICD criteria and were fasting for at least 12 h before collection of blood
The concentration of intestinally derived chylomicrons is indicated as apo B48, an obligatory component of these macromolecules. We found that when fasted for 12 h, the concentration of apo B48 was not significantly different between AD, MCI and control subjects. However, chylomicron homeostasis was also investigated in postabsorptive state 4 h after a standard low-fat breakfast. We found that subjects with AD had significantly greater apo B48 than control subjects with a trend towards significance for subjects with MCI (Table 1).
A representative separation of Aβ peptides on which quantitation was based is depicted in Figure 1. The fasting lipoprotein Aβ concentration for the TRL fraction, LDL and HDL is depicted in Table 2 for subjects with AD, MCI and for controls. For all subject groups, the majority of Aβ was associated with the TRL fraction encompassing chylomicrons and their remnants and hepatically derived VLDL/IDL. The mean total lipoprotein-Aβ for the AD group was 35% greater than for controls or MCI subjects, but there was a significant variation within each group.
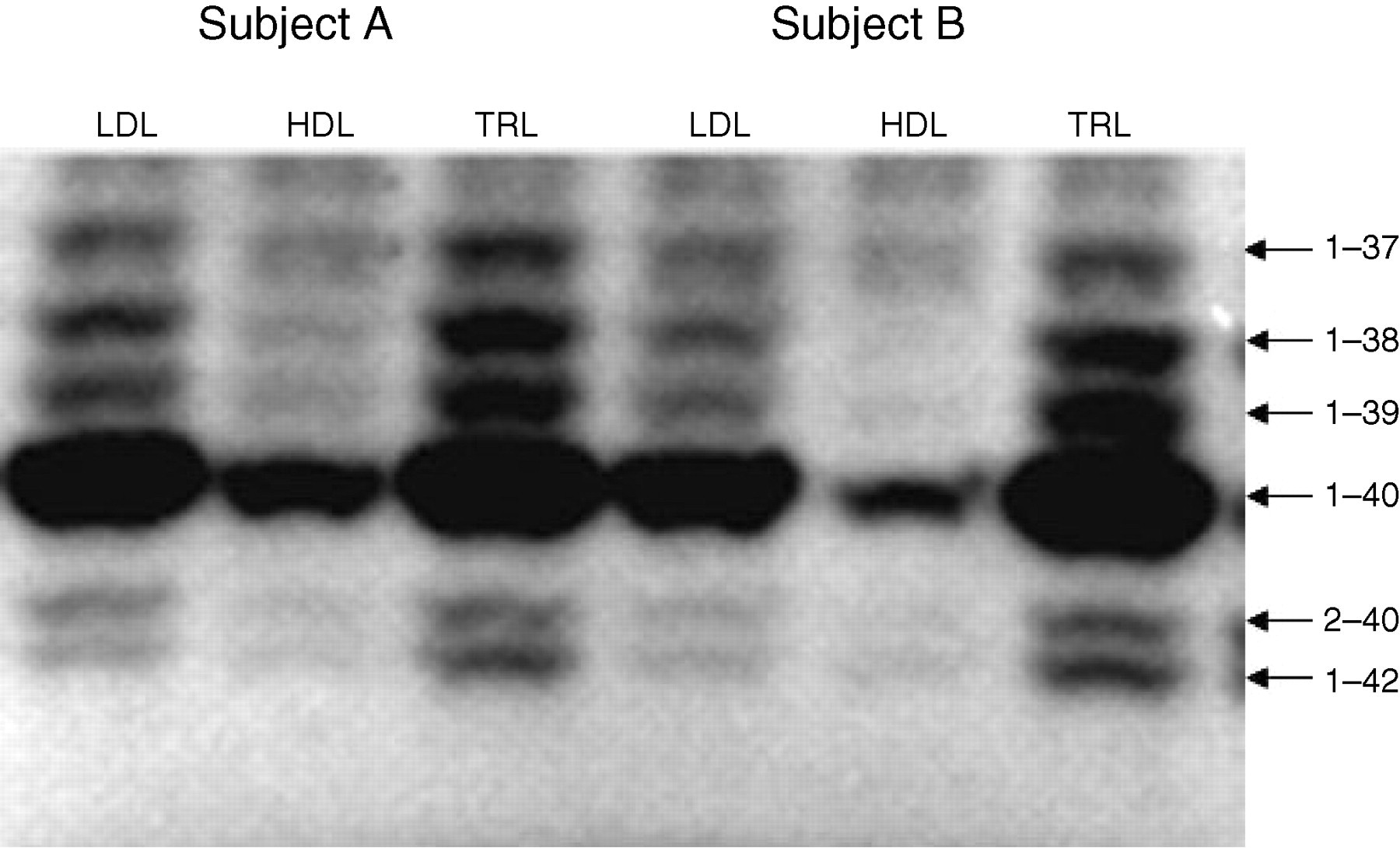
Representative separation of beta-amyloid (Aβ) isoforms (1–37, 1–38, 1–39, 1–40, 2–40, 1–42) in the lipoprotein fractions of two control subjects. Subjects were fasted for 12 h before blood collection and lipoprotein fractions isolated by serial ultracentrifugation. Triglyceride-rich lipoproteins (which includes chylomicrons, VLDLs and their remnants [ρ < 1.109 g/mL]). LDL (1.063–1.019 g/mL) and HDL (1.064–1.21 g/mL). Plasma lipoprotein fractions were immunoprecipitated and separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis. The Aβ isoforms were transferred to membranes and immunostained
The distribution of beta-amyloid (Aβ) in subjects with Alzheimer's disease (AD), mild cognitive impairment (MCI) and control subjects
Subjects were fasted for 12 h before blood collection and lipoprotein fractions were isolated by serial ultracentrifugation. TRLs, triglyceride-rich lipoproteins (which includes chylomicrons, VLDLs and their remnants (ρ < 1.109 g/mL). LDL (1.063–1.019 g/mL) and HDL (1.064–1.21 g/mL)
*TRL-Aβ is significantly different from LDL-Aβ or HDL-Aβ at P < 0.05
To explore whether the lipoprotein distribution of lipoprotein Aβ was interdependent, correlation analysis was determined. We found a significant association of the TRL-Aβ with LDL-Aβ, but not with HDL-Aβ (Figure 2).
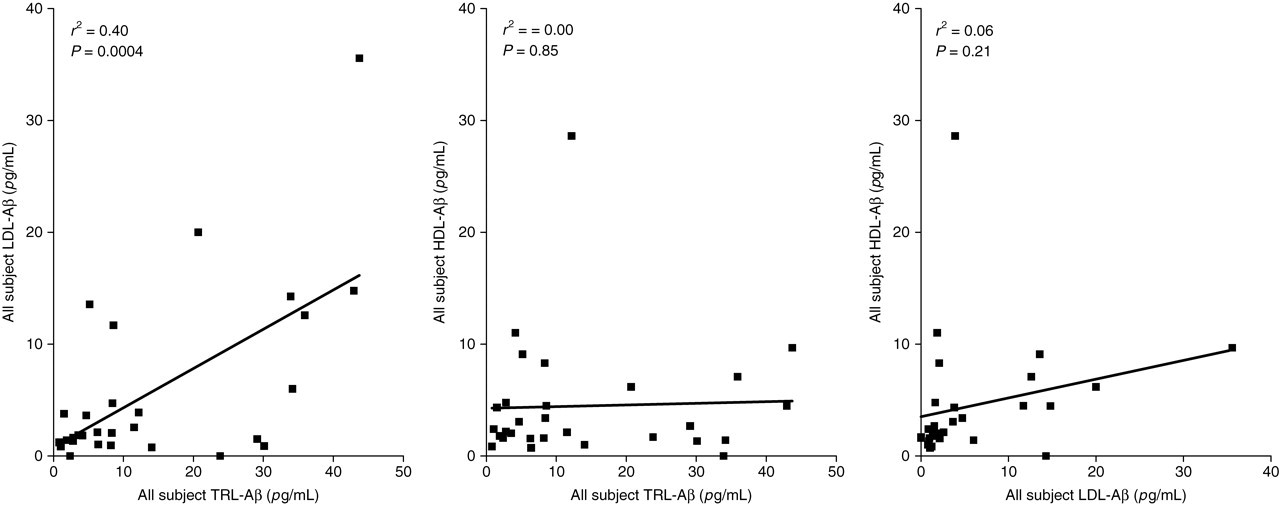
Correlation analysis of beta-amyloid (Aβ) between lipoprotein fractions isolated by serial ultracentrifugation. The triglyceride-rich lipoprotein fraction includes chylomicrons, VLDLs and their remnants (ρ < 1.109 g/mL); LDL (1.063–1.019 g/mL) and HDL (1.064–1.21 g/mL) are indicated. Lipoproteins were isolated from plasma by serial ultracentrifugation. Plasma lipoprotein fractions were immunoprecipitated and separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (Figure 1). The Aβ isoforms were transferred to membranes and immunostained
The Aβ isoforms for TRL, LDL and for HDL in all subjects is shown in Figure 3. For all lipoprotein groups, Aβ1–40 was the predominant isoform accounting for approximately 50% of total Aβ within each lipoprotein fraction. Approximately equivalent amounts of the isoforms 1–42, 2–40, 1–38, 1–37 and 1–39 were found within each lipoprotein group, comprising approximately 10% of total lipoprotein-Aβ.
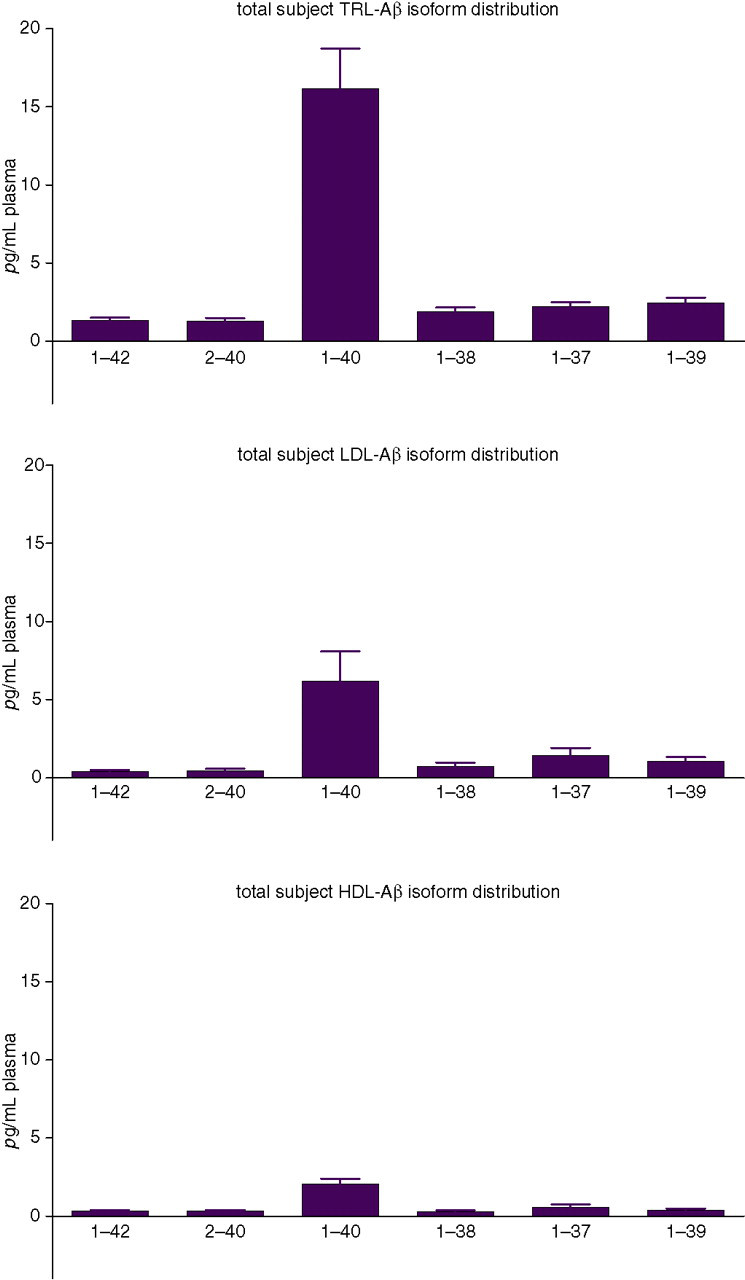
The distribution of beta-amyloid (Aβ) isoforms in the triglyceride-rich lipoprotein fraction (which includes chylomicrons, VLDLs and their remnants (ρ < 1.109 g/mL); LDL (1.063–1.019 g/mL) and HDL (1.064–1.21 g/mL) are indicated. Lipoproteins were isolated from plasma by serial ultracentrifugation. Plasma lipoprotein fractions were immunoprecipitated and separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (Figure 1). The Aβ isoforms were transferred to membranes and immunostained
The distribution of Aβ isoforms within lipoprotein fractions of subjects with AD or MCI and controls is compared in Figure 4. The Aβ1–37, Aβ1–38 and Aβ2–40 isoforms were significantly enriched within the TRL fraction of AD/MCI subjects with 87%, 70% and 56% enrichment, respectively, and similar trends were observed for isoforms Aβ1–39, Aβ1–40 and Aβ1–42.
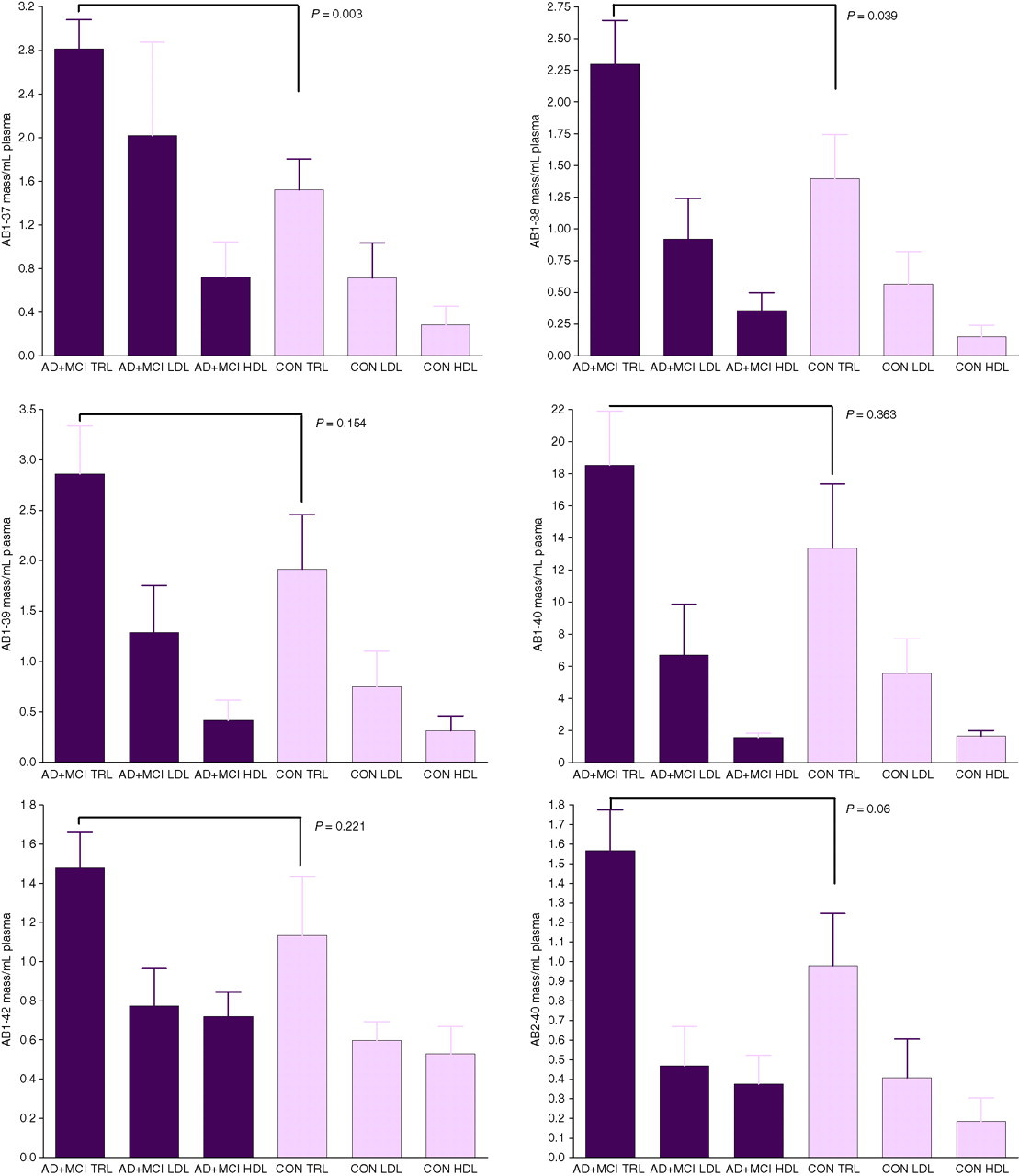
A comparison of beta-amyloid (Aβ) isoforms distributed in plasma lipoproteins isolated from subjects with Alzheimer's disease and mild cognitive impairment vs. age-matched control subjects. The triglyceride-rich lipoprotein fraction includes chylomicrons, VLDLs and their remnants (ρ < 1.109 g/mL); LDL (1.063–1.019 g/mL) and HDL (1.064–1.21 g/mL) are indicated. Lipoproteins were isolated from plasma by serial ultracentrifugation. Plasma lipoprotein fractions were immunoprecipitated and separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (Figure 1) and the Aβ isoforms were transferred to membranes and immunostained
Correlation analysis was done between plasma lipid variables, insulin and BMI with lipoprotein-Aβ, in order to gain insight into potential regulating variables of Aβ distribution. Total plasma cholesterol was negatively associated with plasma total lipoprotein-Aβ, TRL-Aβ, LDL-Aβ and HDL-Aβ (Table 3). The cholesterol/lipoprotein-Aβ negative association was evident with individual Aβ isoforms and most significantly for Aβ1–40, Aβ1–42 and Aβ2–40. When compared with the isolated lipoprotein fractions, total cholesterol showed a negative correlation with the majority of the LDL-Aβ isoforms and also with all isoforms of HDL-Aβ, but no association was found for TRL-Aβ isoforms. We found that the negative association of plasma cholesterol with total lipoprotein-Aβ and with isoforms of Aβ in the LDL and HDL fractions was essentially mirrored for LDL-cholesterol and plasma Aβ distribution (Table 3).
Correlation analysis between plasma total apoprotein (apo) B, plasma cholesterol and LDL cholesterol with plasma beta-amyloid (Aβ) associated with triglyceride-rich lipoproteins (TRLs), LDL and HDL
The numbers 1–42, 2–40, 1–40, 1–38, 1–37 and 1–39 indicate the amino acid isoforms of Aβ determined. Data indicate correlation value (r 2) with negative numbers being indicative of an inverse relationship. Probability of a significant correlation is indicated for P values better than 0.05
Total apo B depicts particle concentration of the TRLs and their remnant lipoproteins. Total apo B had a significant negative association with HDL-Aβ isoforms and consequently total HDL-Aβ. However, total apo B did not correlate with Aβ in either of the two apo B-rich lipoprotein fractions isolated (i.e. TRL and LDL).
While the majority of plasma Aβ was found to be associated with plasma TRLs, there was no evidence based on the correlation analysis that triglyceride per se was a regulating factor of Aβ concentration or distribution (data not shown).
Discussion
In this study, we have investigated the distribution of Aβ in the plasma lipoprotein fractions from subjects with AD and MCI. We found the majority of Aβ to be associated with the TRLs encompassing chylomicrons and their remnants, VLDL and IDL. The Aβ1–37, Aβ1–38 and Aβ2–40 isoforms are significantly enriched within the TRL fraction of AD/MCI subjects and similar trends are observed for isoforms Aβ1–39, Aβ1–40 and Aβ1–42. Plasma total- and LDL-cholesterol are found to be inversely associated with lipoprotein-Aβ. Moreover, there was a particularly evident inverse relationship between plasma cholesterol and isoforms of Aβ within LDL and HDL. While total apo B showed a uniform strong negative association with HDL-Aβ isoforms (and net HDL-Aβ), this was not observed for either LDL- or TRL-Aβ. Collectively, our data shows that Aβ is commonly found associated with plasma lipoproteins, especially those relatively enriched with triglyceride. We find that isoforms of Aβ may be increased in otherwise normolipidaemic AD/MCI subjects and that differences in distribution are evident when compared with controls. The inverse association of both cholesterol and apo B with Aβ concentration and distribution provides possible clues as to the origin and kinetics of lipoprotein-Aβ.
The common association of Aβ with lipoproteins suggests that like other apoproteins, a physiological role of Aβ may be in the regulation of lipid metabolism. Koudinov 31 showed that Aβ is secreted by HepG2 cells as an apolipoprotein complexed with apoA-I, apoJ and transthyrethin lipids, and in other studies showed that Aβ1–40 inhibits lipid biosynthesis. 32 We extend upon those findings and now show a relatively greater plasma distribution of Aβ with the plasma TRLs. While it is not possible to discriminate between TRLs of intestinal and hepatic origin, it is probable that in the fasted state hepatically derived VLDL is the predominant source of plasma Aβ.
The observation that TRL-Aβ correlates with the lipid-depleted remnant lipoproteins raises the possibility that Aβ is involved in the conversion of lipoproteins to the high-uptake remnant forms. Consistent with this concept were previous studies, where it was reported that when Aβ1–42 was complexed to chylomicrons, the protein remained with chylomicrons during the catabolic cascade. 25
The concentration of chylomicrons was determined as apo B48. When fasted, there were no aberrations in chylomicron homeostasis in AD or MCI subjects, but in the postabsorptive state, subjects with AD were found to have significantly increased chylomicrons. Note that triglycerides are a poor surrogate marker of chylomicron homeostasis because concentration is primarily dependent on lipolysis, and not removal of the particle. Indeed, we find no difference in triglyceride concentrations between AD subjects and controls at 4 h following the breakfast meal. Physical activity is also known to attenuate postprandial lipaemia, but young healthy subjects have no significant effect on apo B48 concentration. 33 In this study, subjects who are sedentary or had undertaken substantial physical activity are excluded. Moreover, based on the activities reported for three days prior to blood sampling, there is no obvious difference in physical activity between subject groups. Elevated apo B48 is consistent with postprandial chylomicronaemia, 34 an exaggerated but transient rise in chylomicrons that occurs following the ingestion of dietary fats. On this basis, we would suggest that subjects with AD may have greater exposure to TRL-Aβ in response to high-fat diets. Enterocytic Aβ is enhanced in animals given high-fat diets, consistent with the possibility that chylomicron production is increased. 24 Alternatively, increased apo B48 in the postabsorptive state may be due to reduced high-affinity clearance of remnant lipoproteins, although this seems unlikely given that AD subjects did not have increased concentrations of total apo B (predominanty of hepatic origin). Chylomicrons and hepatically derived apo B lipoproteins utilize the same pathways of removal.
Proteolytic processing of the Aβ precursor protein by beta- and gamma-secretases generates Aβ peptides. The cleavage by secretases occurs predominantly in postgolgi secretory compartment and postgolgi endocytic compartment and is influenced by cholesterol, indicating a role of the membrane lipid composition in proteolytic processing of the Aβ precursor protein. 35 During the genesis of primordial TRLs, apoproteins are complexed to lipids and it is possible that Aβ associated at this point reflects a separate pool of Aβ than that sequestered at the surface or in the vicinity of the plasma membrane by other chaperone proteins. Immunohistochemical studies from our laboratory are consistent with this notion. We found strong staining for Aβ within the perinuclear region of absorptive cells of the small intestine, the exact site of chylomicron assembly. 24 Furthermore, in this study LDL-Aβ correlated positively with TRL-Aβ, which we interpret as being indicative of a product to precursor relationship. In contrast, no relationship between TRL-Aβ and HDL-Aβ was found, excluding the likelihood that the protein is shed to nascent HDL during the lipolytic cascade. While TRLs do not interact substantially with the plasma membrane until hydrolyzed, HDLs do, primarily through the binding cassette protein, ABCA1. It is possible that the HDL accumulates Aβ as a consequence of colocalization with processing of the precursor protein at the plasma membrane.
An increase in cholesterol concentration in neuronal membranes may facilitate the generation and aggregation of the Aβ 36 and high concentrations of cholesterol are associated with an increased risk of AD. 37,38 On the other hand, many studies do not support elevated cholesterol concentrations in serum and brain as risk factor for AD. 39 The beta-secretase, BACE, is a membrane-spanning aspartic protease, which cleaves amyloid precursor protein (APP) in the first step of proteolytic processing leading to the formation of the neurotoxic Aβ. The regulation of the beta-secretase and of BACE access to APP is lipid-dependent, and involves lipid rafts. Several groups of lipids including sterols have been shown to stimulate BACE, 40,41 supporting the view that a reduction of brain cholesterol could be protective against AD. However, genetic and pharmacological evidence has found that low brain cholesterol leads to neurodegeneration, suggesting that the loss of neuronal membrane cholesterol contributes to excessive amyloidogenesis. 42 Reconciliation of the cholesterol/Aβ paradox is provided by the findings of several groups. Galbete et al. 43 reported that cholesterol decreases the secretion of APPs by interfering with APP maturation and inhibiting glycosylation of the protein. They found that although APP is inserted in the membrane it is not cleaved by secretase. McLaurin et al. 44 reported that membrane cholesterol inversely correlated with the extent of Aβ1–40 and Aβ1–42 bilayer interaction and Gibson-Wood et al. 45 found that distribution of cholesterol rather than the total mass is what regulates Aβ genesis. Racchi et al. 46 showed that membrane sterol enrichment by incubation with cholesterol-rich β-VLDL caused a dose-dependent inhibition of Aβ release. In transgenic mice expressing the Swedish familial AD mutations, changes in concentrations of sAPP and Aβ in brain were all negatively correlated with serum cholesterol concentrations. 47 ABCA1 promotes cholesterol efflux from cells and is required for maintaining plasma cholesterol concentrations. In a study by Burns et al. 18 overexpression of ABCA1 achieved by stimulating liver X receptors increased plasma cholesterol and apo E, commensurate with a decrease in brain Aβ.
In this study, we found a strong inverse relationship of both plasma- and LDL-cholesterol with lipoprotein Aβ, which based on the studies cited 39–47 would lead to a reduction in APP processing by secretases. The putative sterol-inhibitory effect on APP processing contrasts with the clinical findings of Kuo et al. 48 who found positive correlations between the concentrations of serum total cholesterol, LDL cholesterol and apo B to the amount of Aβ1–42 in AD brains of human subjects postmortem, but not to Aβ1–40. However, raised cholesterol may increase plasma Aβ concentration and exacerbate cerebral amyloidosis through other mechanisms. Cholesterol-rich diets suppress clearance of apo E-rich remnant lipoproteins by high-affinity pathways and via heightened inflammatory processes. 49
The inverse but strong association of total apo B with HDL-Aβ would seem surprising given that there was no relationship between TRL-Aβ and HDL-Aβ, nor LDL-Aβ and HDL-Aβ. However, these observations are consistent with the suggestions that Aβ is not transferred between lipoproteins, but rather is regulated by cellular cholesterol homeostasis. The majority of total apo B is found as LDL; hence a higher apo B is indicative of greater plasma cholesterol concentrations and therefore by extrapolation of lower rates of Aβ biogenesis.
The total lipoprotein-Aβ concentration was on average approximately 45% greater in AD subjects when compared with controls; however, there was significant variability within each group. Nonetheless, of the 11 subjects who had a TRL-Aβ above the group mean, we found that nine were AD or showed MCI. The tendency for greater total-Aβ in AD subjects was predominantly reflected in the apo B-containing lipoproteins relative to HDL, the latter comprising just 13% of the plasma lipoprotein total-Aβ. Significantly greater quantities of Aβ1–37, Aβ1–38 and Aβ2–40 were found in the AD/MCI group when compared with controls and there was a trend towards significance for other Aβ isoforms. We extend on the findings by Smith and Betteridge 50 who showed that dyslipidaemic subjects had significantly greater plasma Aβ1–40 and report that the concentration of Aβ isoforms, particularly those bound to LDL and HDL, inversely correlated with total cholesterol or LDL cholesterol.
The origin of Aβ fibrillar deposits in senile plaque is not unequivocally established and indeed may be multifactorial. Dysregulation in Aβ production, sequestration or delivery/efflux across the BBB may contribute to cerebral amyloidosis. Studies by Reese and Karnovsky 51 and Brightman 52 demonstrated that brain endothelial cells prevent trans-capillary movement of polar molecules and found no detectable trans-endothelial pathways. However, not all areas of the brain contain capillaries that produce a barrier. In these no-barrier regions, the morphological features of the capillaries are similar to those of systemic microvascular beds. Thus, the tight junctions are discontinuous, there are more plasmalemmal vesicles and some endothelial cells exhibit fenestrations. Animal model studies demonstrate that the BBB may become compromised with age. Lutjohann et al. 53 showed that the concentrations of the exogenous plant sterols campesterol and sitosterol were significantly elevated in the brains of APP23 animals at 12 and 18 months of age, coinciding with abundant plaque formation. Furthermore, apolipoproteins B100 and B48, equivocal markers of hepatic and intestinal lipoproteins, respectively, have been reported in cerebrospinal fluid of otherwise normal healthy subjects. 54–56
Apo E-rich TRLs have high affinity for extracellular matrices and serve as substrate for inflammatory cells, eliciting a respiratory burst that can compromise cell viability and indeed lead to cell death. 57 Although evidence is presently lacking, it is possible that amyloid deposits in senile plaques are, in part, a reflection of exaggerated systemic delivery of lipoprotein-associated Aβ.