
Abstract
A 20-year-old fit male soldier presented on two separate occasions 16 months apart with severe, symptomatic hyponatraemia and a clinical and biochemical picture consistent with the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). In the intervening period, repeated plasma sodium values were in the reference range. Intensive investigation failed to reveal a cause for SIADH that was initially considered idiopathic. The description of a family comprising several adults with intermittent or water load induced-hyponatraemia associated with an activating mutation in the arginine vasopressin (AVP) receptor type 2 (AVPR2) raised the question of whether our patient could have a similar ‘nephrogenic syndrome of inappropriate antidiuresis’. Mutational screening of AVPR2 in our patient revealed a single missense mutation (R137C) in the second intracellular loop, which has been associated with constitutive activation of the AVPR2. In conclusion, adults with intermittent, severe hyponatraemia may have a constitutively activating mutation in the AVPR2 with resultant nephrogenic syndrome of inappropriate antidiuresis. Patients with idiopathic SIADH, particularly those with unmeasurable circulating AVP concentrations, should be considered for mutational screening of AVPR2.
Introduction
The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) is the most common cause of hyponatraemia and is defined by the combination of decreased plasma osmolality (<275 mOsm/kg), inappropriately concentrated urine with osmolality > 100 mOsm/kg and inappropriate natriuresis (urine sodium > 40 mmol/L) in a clinically euvolaemic patient with normal thyroid, renal and adrenal function. 1 Although the majority of patients with apparent SIADH have detectable circulating arginine vasopressin (AVP) despite reduced plasma osmolality, as many as 10–20% of affected patients have AVP concentrations at or below the limits of detection by radioimmunoassay. 2 Until recently, the mediator(s) of the urinary concentration in this group of patients has remained elusive.
The description in 2005 of an activating mutation in a highly conserved region of the X-linked AVP receptor type 2 (AVPR2) in two hemizygous male infants with persistent severe hyponatraemia and undetectable circulating AVP provided a clear explanation for urinary concentration without detectable AVP in these infants. 3–5 The so-called nephrogenic syndrome of inappropriate antidiuresis was subsequently described in a five-generation family with three hemizygous adult males and four heterozygous adult females. 6 Spontaneous hyponatraemia or an abnormal water load test were noted in six of the seven mutation-positive subjects with the only phenotypically unaffected female having evidence of skewed X inactivation with preferential inactivation of the mutated allele. We now describe an adult male with intermittent, severe and symptomatic hyponatraemia due to SIADH who was found to have the previously reported activating mutation in the AVPR2. This is only the second report of an adult with the nephrogenic syndrome of inappropriate antidiuresis and emphasizes that the hyponatraemia may be intermittent and severe, while also raising the possibility that the prevalence of this condition may be higher than initially considered.
Case report
A 20-year-old previously healthy male soldier was referred to the Endocrinology Department for evaluation of severe hyponatraemia. The evening of his return to New Zealand from deployment in the tropics, he visited a bar and had two standard beers. He awoke the following morning with a slight headache and nausea and then had a generalized seizure. There was no history of exposure to a thiazide diuretic, selective serotonin reuptake inhibitor (SSRI) antidepressant or 3,4-methylenedioxymethamphetamine (MDMA) (‘ecstasy’). 7 On admission, he was clinically euvolaemic with a blood pressure of 115/64 mmHg and although postictal had no focal neurological signs. Plasma sodium was 120 mmol/L, potassium 4.1 mmol/L, glucose 5.1 mmol/L, urea 2.9 mmol/L and creatinine 70 μmol/L. Plasma osmolality was 244 mmol/kg (280–300), urine osmolality 579 mmol/kg with urine sodium 117 mmol/L. He was biochemically euthyroid with thyroid-stimulating hormone 1.37 mIU/L (0.25–2.5) and free T4 index 90 (55–160). A Synacthen test was normal with a basal cortisol of 429 nmol/L and +30 min value of 686 nmol/L (normal > 550). An overnight metyrapone test was also performed to exclude partial adrenocorticotrophic hormone (ACTH) deficiency and was normal with post-metyrapone 11-deoxycortisol 509 nmol/L (>220) and ACTH 78.8 pmol/L (>30). A diagnosis of SIADH was made and further investigations to determine a cause, including chest X-ray and brain magnetic resonance imaging, were normal. He was treated with fluid restriction and his plasma sodium gradually increased to 127 mmol/L on day 4 and had normalized by day 7. A further nine plasma sodium values over the next 16 months ranged from 135 to 142 mmol/L. A diagnosis of transient SIADH of uncertain cause was made.
Sixteen months after his initial presentation he was transferred back to the tropics for active military duty. Within days of arriving, he exercised for about 4 h in an ambient temperature of approximately 35°C and recalls consciously drinking pure water in excess of his thirst. He felt lethargic at the end of the session and the following morning had a headache. Plasma sodium was checked and found to be 117 mmol/L. He was hospitalized, treated with intravenous saline and oral salt supplementation and his physical duties were restricted – his sodium however remained in the borderline at 130–134 mmol/L for several weeks. On his return to New Zealand three months later, further investigations were arranged. In view of the documented association between cystic fibrosis and hyponatraemic heat exhaustion, a sweat test was performed and gave normal results with 27 mmol/L chloride (normal < 40). 8 No samples had been assayed for AVP at the time of hyponatraemia. The possibility of an activating mutation in the AVPR2 was then considered as an explanation for his intermittent, severe hyponatraemia.
After genomic DNA extraction, all three coding exons of the V2R (AVPR2) gene were amplified using the polymerase chain reaction (PCR) and analysed by automated bidirectional fluorescent DNA sequencing. Sequence was compared with the GenBank reference UO4357 and confirmed that the patient was hemizygous for the V2R gene mutation c.409C > T, resulting in the substitution of the normal arginine for a cysteine residue at amino acid position of the vasopressin V2 receptor (p.Arg137Cys) (Figure 1). This is a previously reported 3 gain-of-function mutation that can cause constitutive activation of the receptor.
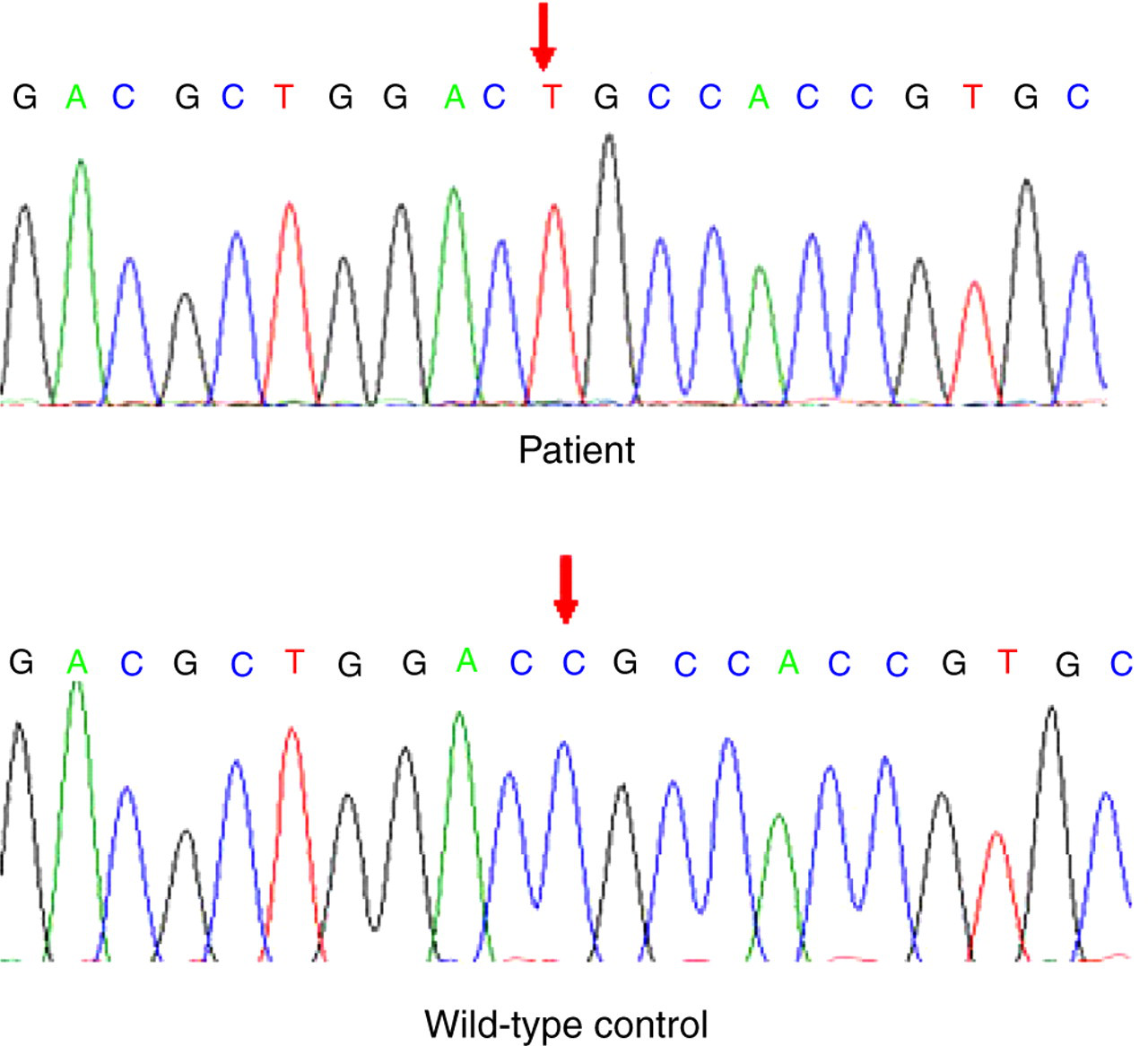
Arginine vasopressin receptor type 2 gene sequences from wild-type control (bottom panel) showing normal CGC sequence, coding for arginine at codon 137 and the mutant sequence from the patient (top panel) with the TGC sequence, coding for cysteine (R137C)
This missense mutation in the second intracellular loop, near the cytoplasmic boundary of the third transmembrane domain, occurs in a highly conserved motif in G protein-coupled receptors and has been associated with a four-fold increase in basal cAMP production via constitutive activation of the AVPR2. 3,6 A final diagnosis of the nephrogenic syndrome of inappropriate diuresis secondary to an activating mutation in the AVPR2 was made. A water load test was considered but in view of the previous episodes of severe hyponatraemia, including one generalized seizure, and the detection of a disease-associated mutation which has clearly been demonstrated to constitutively activate AVPR2, this was not performed. Family screening was not possible as he is adopted and has no contact with his birth relatives. He has been advised to avoid strenuous exercise in hot and humid conditions, to have regular assessments of plasma sodium concentrations if exposed to high ambient temperatures and to avoid excessive hypotonic fluid intake while exercising.
Discussion
SIADH, first described by Bartter and Schwartz in 1967, is a common complication of a wide variety of clinical conditions and drug therapies and is characterized by hyponatraemia secondary to increased total body water and decreased total body sodium. 9,10 Shortly after the description of the syndrome, it was recognized that 10–20% of patients with SIADH had unmeasurable circulating concentrations of AVP despite hyponatraemia with inappropriately concentrated urine. 2,11 The mediator(s) of the urine concentration in this group of patients remained elusive until the description of two unrelated male infants with chronic moderately severe hyponatraemia (plasma sodium 118 and 123 mmol/L) both hemizygous for a specific constitutive activating mutation in the X-linked AVPR2. 3 The only report to date in adults described a large pedigree with the previously documented activating R137C mutation in AVPR2 in three hemizygous males and four heterozygous females. 6 There was a high degree of phenotypic variability noted with six of the seven mutation-positive subjects manifesting either intermittent hyponatraemia or normonatraemia with abnormal excretion of water load. Of note, several of the AVPR2 mutant subjects had intermittent hyponatraemia that seemed to depend on reported fluid intake.
The patient we describe is only the second report of an adult with hyponatraemia secondary to an activating mutation in AVPR2 and extends the range of possible presentation of patients with this presumably rare disorder. The possibility of this disorder was considered, despite the fact that AVP had not been measured at the time of hyponatraemia, although it would have been expected to be low or undetectable. The point of particular interest is the intermittent and severe nature of the hyponatraemia demonstrated in our patient. The explanation for the first episode of hyponatraemia remains elusive. The possibility of exposure to a drug such as MDMA was considered, but this was denied by the patient (but unfortunately not screened for). There was no good support for excessive fluid ingestion and no obvious trigger to AVP secretion, such as pain or nausea.
The second episode of hyponatraemia occurred in the setting of prolonged exercise shortly after arriving in a tropical environment, a situation clearly associated with the development of hyponatraemia in susceptible individuals. 12 More than 100 cases of exercise-associated hyponatraemia have been reported from physical activities as diverse as military marches, prolonged hiking and marathon races with at least five documented fatalities attributable to hyponatraemia. 13 A study of runners in the Boston marathon revealed that 13% were hyponatraemic on completion of the race, while 0.6% had severe hyponatraemia (sodium < 120 mmol/L). 14 The major independent determinants of hyponatraemia were weight gain and a racing time of >4 h, supporting previous suggestions that dilutional hyponatraemia secondary to excess fluid ingestion during exercise was the major pathophysiological factor. 15 However, there is also evidence supporting inappropriate antidiuresis as a contributing factor in exercise-associated hyponatraemia. Seven of 44 (16%) Grand Canyon hikers were hyponatraemic with plasma sodium 109–127 mmol/L, and all had documented fluid intakes greater than normonatraemic hikers. Of particular interest, urine osmolality in these hyponatraemic hikers ranged from 476 to 609 mOsm/kg, suggesting a contribution of inappropriate antidiuresis to the hyponatraemia. 16 Our patient exercised in a tropical environment immediately prior to his second episode of hyponatraemia. Potential contributors to the development of the acute, severe hyponatraemia on this occasion would include sweating with associated increased loss of sodium, hypotonic fluid replacement and finally exercise-associated AVP release (mediated by increased body temperature, stress and possibly nausea) in the setting of a patient with a constitutively activated AVPR2-limiting free water excretion. 12,17 It is possible that certain individuals may be at particularly high risk of severe exercise-associated hyponatraemia, with one potential contributor being activating mutations or polymorphisms of AVPR2.
The mainstay of treatment for patients with the nephrogenic syndrome of inappropriate antidiuresis is fluid restriction, although the degree of fluid restriction required depends on the severity and chronicity of the hyponatraemia. Our patient has been advised to avoid excessive hypotonic fluid intake during exercise and to ensure that plasma sodium is measured if he develops any symptoms compatible with hyponatraemia. Agents that inhibit AVPR2 signalling downstream of AVP binding (lithium and demeclocycline) could be considered, although the intermittent nature of his hyponatraemia and the toxicity of these agents argue against their use in our patient. AVPR2 competitive antagonists are not useful in the presence of a constitutively activated receptor – indeed, the first reported adult case of nephrogenic inappropriate antidiuresis was detected following the observation of a failed response to two oral inhibitors of AVPR2. 6,17 However, an agent that functions as an inverse agonist and stabilizes the receptor in an inactive conformation has been described and may ultimately become an ideal treatment for this condition. 18
In conclusion, adults with hyponatraemia may have a constitutively activating mutation in the AVPR2 with resultant nephrogenic syndrome of inappropriate antidiuresis. The hyponatraemia in these patients may be severe and intermittent, presumably related to variations in the degree of sodium depletion, hypotonic fluid intake and AVP secretion. This report emphasizes that mutational screening of the AVPR2 should be considered in patients with both intermittent and sustained, apparent SIADH, particularly if associated with an undetectable circulating AVP concentration. Since up to 20% of patients with SIADH have unmeasurable plasma AVP, this syndrome may be more common than initially anticipated and further studies exploring this hypothesis are warranted.