
Abstract
Abstract
Obesity is now regarded as a global epidemic affecting both adults and children, and is associated with significant morbidity and mortality. Thus the effective management of obesity has become an important clinical focus. Therefore, an understanding of the pathways controlling appetite, satiety and food intake is critical for gaining an insight into the pathogenesis of obesity and also for the development of diagnostic tests and therapeutic agents for use in the clinical management of this condition. Over the last decade or more research using both mouse and human genetic models has elucidated the critical role of the leptin-melanocortin pathway in the hypothalamus, in regulating mammalian energy balance. In tandem with this, a clearer understanding of the regulation of gut-derived hormones and their interaction with the central nervous system has further illuminated the complex interplay between central and peripheral aspects of energy regulation. The obesity epidemic and the expanded knowledge base relating to its aetiopathogenesis have specific implications for clinical biochemistry. In particular, an increase in workload may be expected due to biochemical investigation of obesity and its co-morbidities. Moreover, advice on the in-depth investigation of complex cases of obesity may be sought, including information on newer diagnostic tests, such as serum leptin or molecular genetic analysis. There may also be a substantive role for chemical pathologists in establishing and running clinical obesity services. Finally, clinical biochemistry has a role in research pertaining to obesity and cardiometabolic risk.
Introduction
It is now almost universally acknowledged that obesity is a major leading public health issue. In the USA, over 60% of adults are now considered overweight and over 30% are clinically obese. 1 Moreover, the prevalence rates in many European countries, including the UK and Ireland, are rapidly approaching the levels encountered in the USA. 2 In general, it is suggested that up to 20% of the population of most Western countries are obese, with up to 50% classified as overweight. Moreover, this problem is not only confined to Western societies and the World Health Organization (WHO) has proclaimed obesity to be a global epidemic. 3
While there are obvious cosmetic and quality-of-life issues related to obesity, the primary focus in the realm of health care is on the substantial morbidity and mortality associated with this condition (Table 1). Clinical problems range from mechanical, such as osteoarthritis and sleep apnoea, to metabolic perturbations resulting in type 2 diabetes (T2DM), hyperlipidaemia, hypertension and an overall increased cardiovascular risk. 4 In addition, there is an increasing realization that obesity is associated with a higher risk for developing certain cancers, e.g. endometrial, colon and prostate. 5
Health consequences of obesity
The marked increase in obesity prevalence has prompted a number of government-led reviews of this health problem in many countries, with the intended remit of preventing the occurrence of obesity and also developing effective clinical management strategies to tackle the clinical ramifications of this disease. 6,7 Such initiatives will have important downstream implications for many areas of the health-care sector, including laboratory medicine.
Measurement of obesity
There are many different methods applied to the classification of obesity and the measurement of adiposity. The WHO classification, which is the most widely applied in clinical practice, is based on the measurement of body mass index (BMI) (weight/height 2 ). 8 In this system, overweight is defined as BMI ≥25 kg/m2, while obesity is determined by BMI ≥30 kg/m2 (Table 2). However, there are caveats in relation to universal application of this classification because of differences observed in the relationship between morbidity and BMI in various ethnic populations. Thus, attempts are being made to adopt population or ethnicity specific classifications of overweight and obesity using BMI.
World Health Organization classification of weight using body mass index (BMI) (1995)
Another limitation of BMI is that it is not a specific descriptor for the distribution of adipose tissue. This has important clinical implications in view of the fact that compelling evidence now indicates that accumulation of visceral adiposity has a particularly strong association with the development of metabolic complications such as T2DM, hypertension and cardiovascular disease (CVD). 9 This aspect of adverse fat distribution is well represented by measurement of waist hip ratio or waist circumference (WC). Notably, the WHO has produced a gender-specific classification for visceral adiposity based on WC, and WC is included in the list of National Cholesterol Education Programme (NCEP) III criteria for metabolic syndrome (Table 3). 10
Diagnostic criteria for the metabolic (insulin resistance) syndrome
The presence of three or more of these factors defines a subject as having metabolic syndrome. WC, waist circumference
Other methods for the measurement of adiposity include anthropometry (multisite skin-fold thickness), bioelectrical impedance, imaging modalities such as magnetic resonance imaging (MRI) and dual energy X-ray absorptiometry (DEXA) scanning and isotopic tracer measurements. 11 These techniques are likely to be available in specialist obesity outpatient clinics and research facilities.
Childhood obesity
A serious consequence of the current obesity epidemic is the alarming increase in the prevalence of childhood-onset obesity. 12 In addition, co-morbidities such as T2DM, and features consistent with the metabolic syndrome, e.g. low plasma HDL-cholesterol, are now emerging in this cohort. 12 The phenomenon of childhood obesity is a portent to a potential health-care deluge, which will severely impact in the future on already overstretched health service resources in many countries.
In children, different criteria are applied to the definition of overweight and obesity. It is recommended that a BMI >95th percentile represents obesity, while a BMI >85th percentile is classified as overweight. 13 Population-specific BMI percentile charts should be derived for this purpose.
Aetiopathology of obesity
A number of factors are thought to contribute to the dramatic increase in the prevalence of obesity observed over the last two decades (Table 4). There has been an increase in the per capita average energy supply worldwide, and the trend for the inclusion in modern diets of food with greater saturated fat and sugar content leads to increased caloric intake. 14 Lifestyle changes, including the adaptation of a more sedentary lifestyle due to changes in working and leisure time demands, are also contributing factors. 15 The increasing reliance on motorized transport relative to cycling and walking is another potential factor. 15,16 In general, there are many other more subtle behavioural and environmental influences that contribute to this ‘obesogenic’ milieu. 15
Aetiology of human obesity
There is also now compelling evidence of a role for genetic influences on body fat mass and BMI. 17,18 Extensive family, twin and adoption studies all support the notion that adiposity is a highly heritable trait and direct estimates of heritability vary from 40 to 70%. 17 Thus, the paradigm that should be applied in relation to common or polygenic obesity is that it is a phenotype which results primarily from gene–environment interactions.
There are some circumstances where obesity has a clear organic or genetic origin. 19 Thus, many commonly used drugs, e.g. insulin, corticosteroids, can increase body weight. Many endocrine and neuroendocrine diseases are associated with weight gain, e.g. Cushing's syndrome, hypothalamic disease and injury. Certain pleiotrophic genetic syndromes, e.g. Prader-Willi, Alstrom syndrome, may have obesity as a feature, and in the last decade the emergence of specific monogenic syndromes of obesity have been reported. 20
The control of appetite and satiety
In simple thermodynamic terms, obesity can be considered as a chronic disorder of energy imbalance, in which a long-term excess of energy intake over expenditure leads to the storage of that excess energy as adipose tissue. 21 Therefore, a central focus in the effort to elucidate the aetiopathogenesis of obesity and weight gain is the attempt to illuminate the complex neuromolecular mechanisms which underlie the regulation of mammalian energy homeostasis.
The role of the hypothalamus in energy homeostasis
The critical role of the hypothalamus in regulating energy intake was finally established over 50 years ago with the classic experiments of Hetherington and Ranson, 22 who placed electrolytic lesions in the rat hypothalamus. Affected animals developed a ‘condition of marked adiposity’. These elegant experimental systems served as a basis for investigating the neural networks operating in the relevant areas of the hypothalamus for many subsequent years. More recently, the delineation of specific molecular genetic defects in well characterized, naturally occurring rodent genetic models of obesity has also facilitated the development of a comprehensive hard wire neural map of the appetite and satiety regulatory pathways within the mammalian central nervous system (CNS) (Figure 1). 23–28 Furthermore, the identification of orthologous genetic defects in human subjects with severe obese phenotypes has provided bona fide evidence of the importance and relevance of these pathways in humans. 29–33
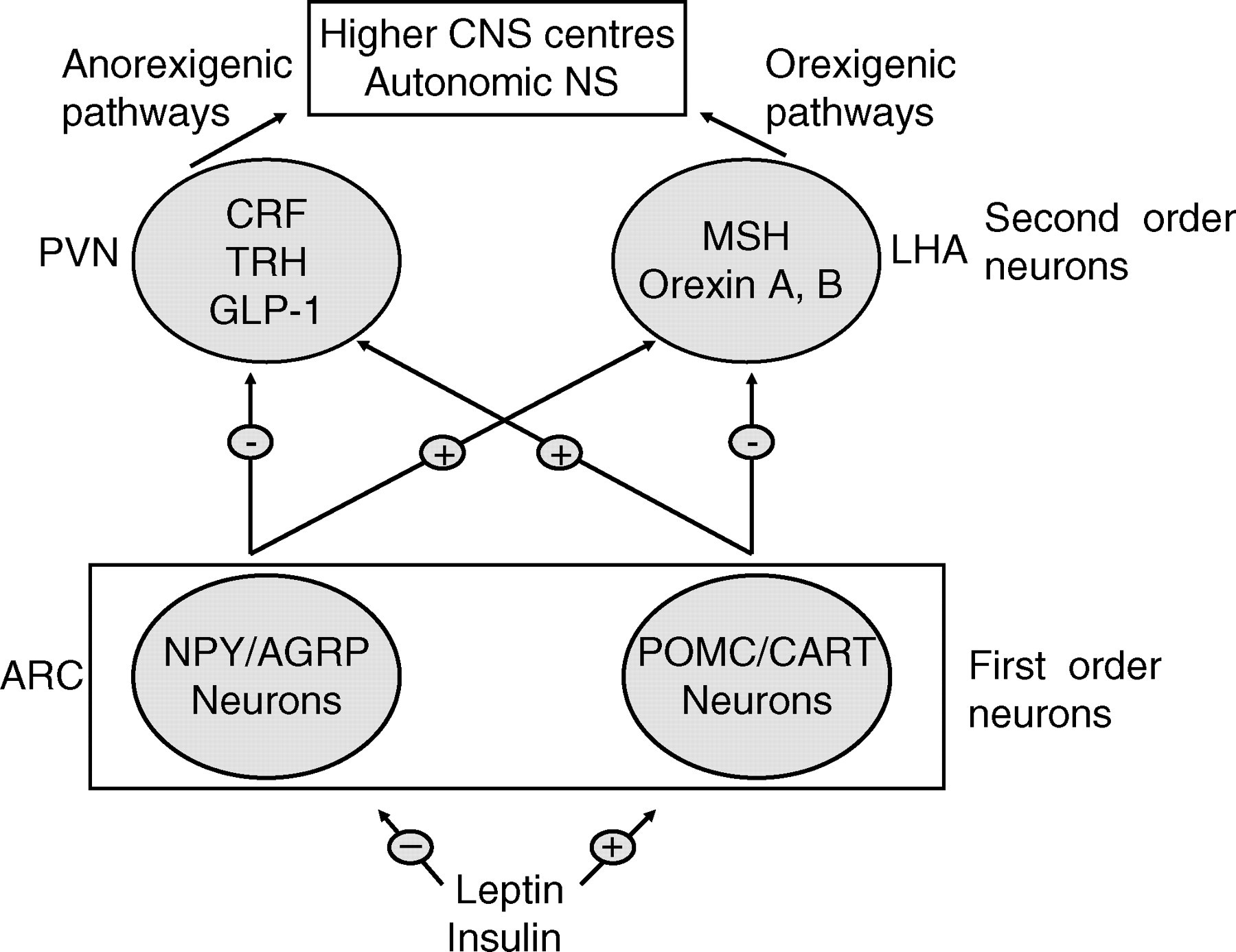
Hypothalamic networks implicated in the control of food intake. AGRP, Agouti-related protein; ARC, arcuate nucleus; CART, cocaine and amphetamine regulated transcript; CRF, corticotrophin releasing factor; GLP-1, glucagon-like peptide-1; LHA, lateral hypothalamic area; MSH, melanocyte-stimulating hormone; NPY, neuropeptide Y; POMC, proopiomelanocortin; PVN, paraventricular nucleus; TRH, thyrotrophin-releasing hormone; NS, nervous system; CNS, central nervous system
For many years, the existence of a circulating factor linking central hypothalamic appetite regulatory pathways and adipose tissue was proposed. The breakthrough in identifying this lipostatic hormone occurred with the discovery of leptin, and the recognition that leptin deficiency was the basis for obesity observed in the ob/ob mouse, an extensively studied rodent model of obesity. 23 Leptin is an adipocyte-derived hormone secreted in response to an increase in energy stores and thus its concentrations in plasma are directly proportional to the degree of body fat. 34 The primary action of leptin in rodents is to exert a long-acting effect of reducing adiposity by attenuating appetite and increasing thermogenesis. Two forms of leptin receptor (LEPR) have been identified, a long form (OB-RL) which appears to mediate leptin signal transduction, and a short form (OB-RS), which is thought to facilitate transport across the blood–brain barrier. 34,35 OB-RL is a member of the cytokine receptor family, and activates the JAK/STAT signal pathway, which employs Janus kinases (JAKs) and signal transducers and activators of transcriptions (STATs).
Molecular mapping of the effects of leptin on the hypothalamus and other areas of the CNS was performed by studying the expression of immediate early genes, particularly c-fos, after both systemic and intracerebroventricular (ICV) administration of leptin. 36 Leptin directly modulated neuropeptide expression in so-called first-order neurons in the hypothalamic arcuate nucleus (ARC), and these neurons in turn regulated, by synaptic input, neuropeptide expression in downstream neurons (second-order neurons or higher). After extensive study it is now apparent that the primary first-order neuronal targets of leptin (and insulin) action are catabolic proopiomelanocortin (POMC)/cocaine and amphetamine-regulated transcript (CART) neurons, which are stimulated by lipostatic hormones, e.g. leptin, and anabolic neuropeptide Y (NPY)/Agouti-related protein (AGRP) neurons which are suppressed by leptin. 36–38
In the currently accepted neuroanatomical framework, POMC/CART neurons project from the dorsolateral ARC to a number of second-order neurons in locations such as the lateral hypothalamic area (LHA) and the paraventricular nucleus (PVN). In the LHA, they appear to make monosynaptic connections with neurons expressing melanocortin-stimulating hormone (MSH) and orexins A and B, suppressing the expression of these neuropeptides. MSH is a particularly potent orexigenic molecule, and ICV injection into normal rodents leads to a marked increase in food intake. 39 In the PVN, which tends to have a primarily catabolic output (compared with the anabolic output of LHA), POMC neurons interact with second-order neurons expressing thyrotropin-releasing hormone (TRH), corticotrophin-releasing hormone (CRH) and oxytocin. Both TRH and CRH have been demonstrated to enhance energy expenditure while decreasing caloric intake. These effects may be mediated, in part, through the thyroid axis and the sympathetic nervous system.
The NPY/AGRP neurons are concentrated in the ventromedial ARC and also project to the LHA and PVN. 36–38 NPY is a highly abundant peptide in the hypothalamus and has the powerful effect of increasing food intake when administered centrally. 40 Moreover in the ob/ob mouse, the absence of NPY does appear to partially ameliorate the obese phenotype caused by congenital leptin deficiency. 41
Finally, as well as communications within the hypothalamus, the POMC/CART and the NPY/AGRP also communicate directly with neural pathways and clusters in other areas of the CNS, such as the nucleus of the solitary tract (NTS) in the brainstem, which has been implicated as a principal target of short-acting meal termination signals, e.g. mechanical gastric stretch, cholecystokinin (CCK) and glucagon-like peptide-1 (GLP-1). Overall, the regulation of energy balance involves a complex network of multiple interacting neuron populations from distinct areas of the CNS. 36–38
Melanocortin system
A substantial body of both genetic and pharmacological research data clearly implicates the melanocortin system as a nodal point in the control of appetite and energy balance. 42,43 While POMC expression is upregulated in response to leptin and increased energy stores, POMC expression in ARC is reduced in conditions such as fasting or leptin deficiency. 44 POMC is post-translationally cleaved, most probably by prohormone convertase 1 (PC1) into multiple peptides which include α-, β- and γ-MSH. 45 In the CNS, two of the five known melanocortin receptors (MCR), namely MC3R and MC4R, are highly expressed in areas of the brain concerned with the regulation of food intake, e.g. PVN, LHA and autonomic activity. 46,47 The MC4R has a high affinity for the agonists α- and β-MSH, but also has a strong affinity for the naturally occurring antagonist AGRP. Thus, it seems that MC4R is a point in the hypothalamus at which fine tuning of the leptin signal cascade takes place, with the relative balance between agonist (MSH) and antagonist (AGRP) determining the effect transduced. 48 Indeed, the administration of low-dose MC4R antagonists attenuates significantly the capacity of exogenously administered leptin to reduce food intake. 49 In addition, deletion of MC4R gene in mice results in a phenotype characterized by early-onset hyperphagia and obesity. 33 The importance of the melanocortin pathway in promulgating the leptin signal cannot be overstated.
Insights from human monogenic syndromes of obesity
While the extensive study and unravelling of the molecular defects in rodent models of obesity confirmed the critical nature of the leptin-melanocortin system in mammalian energy regulation, it remained to be determined whether the importance of this control pathway could be extrapolated to the more complex biological systems operating in humans (Table 5). Through the identification of a number of monogenic disorders, in which early-onset obesity is a pre-eminent phenotype, the relevance of the leptin-melanocortin pathway to human appetite regulation was firmly established. 29–33,50,51
Monogenic causes of human obesity
Within a few years of the discovery of leptin and its role in the pathogenesis of obesity in the ob/ob mouse model, the first human cases of congenital leptin deficiency were described by O'Rahilly's group in the University of Cambridge, UK. 29 Two severely obese cousins from a highly consanguineous family of Pakistani origin were found to be homozygous for a frameshift mutation (ΔG 133) in the LEP gene. 29 This resulted in the production of a truncated protein, which was not secreted and, thus, despite their obesity, plasma leptin concentrations were paradoxically undetectable in these subjects. 52 Subsequently, four other affected individuals from three different Pakistani kindreds, who are also homozygous for the same mutation, were identified. This suggests either that the mutation is a founder mutation in this ethnic group or that the area of the LEP gene affected, which contains a sequence of six guanines, is a mutation hotspot. A Turkish kindred has also been identified in which a different missense mutation (A105W) is pathogenic for this condition. 53
A clear phenotype associated with human congenital leptin deficiency has emerged. 54–56 Affected subjects are characterized by severe early childhood-onset obesity and marked hyperphagia. In addition, hyperinsulinaemia and advanced bone age are noted, and in adults hypogonadotrophic hypogonadism is a feature, although in one adult subject spontaneous puberty did occur but was delayed. Overall, most of the phenotypes manifest in ob/ob mice are recapitulated in the humans. However, species-specific phenotypic differences were evident in that humans did not present with certain features, including growth retardation, hypercortisolaemia and reduced energy expenditure. 54
Consistent with the observed profound anti-obesity effect of leptin in ob/ob mice, 57 leptin replacement in congenital leptin-deficient subjects also produced a dramatic response. 54,55 Daily subcutaneous injections of leptin substantially reduced body weight, with 98% of weight loss occurring in fat mass, this effect being evident in both children and adult leptin-deficient subjects. The principal action of leptin involved a significant attenuation of hyperphagia and associated caloric intake. One female subject progressed into puberty at the appropriate age, suggesting that leptin replacement facilitated this action. Of note also, while overt central hypothyroidism was not a clinical feature in these patients, plasma-free T4 concentrations showed an increase from the earliest post-treatment time point and appeared to stabilize around a new concentration. This is consistent with the observation that leptin has a role in the regulation of TRH secretion from the hypothalamus. 58 Thus, overall, leptin deficiency represents a form of monogenic obesity, which is directly amenable to mechanism-based therapy.
A pathogenic mutation resulting in LEPR deficiency has been described in a kindred of Algerian origin. 30 Three subjects with severe early-onset obesity were homozygous for a splicing mutation (IVS16 +1 G > A), which truncates the LEPR before the transmembrane domain, thus resulting in an inability to transduce the leptin signal. The phenotype associated with this abnormality was very similar to the ligand (leptin)-deficient state, however, certain neuroendocrine features were noted which appeared unique to LEPR deficiency, including impaired growth hormone secretion with growth retardation, decreased IGF-1 and IGF-BP3 plasma concentrations and overt hypothalamic hypothyroidism. In addition, markedly elevated leptin concentrations were observed, but this may, in part, represent an effect related to high concentrations of circulating leptin bound to the truncated LEPR rather than ‘leptin resistance’ per se. Recently, a comprehensive report of the phenotype associated with inherited LEPR deficiency indicated that normal concentrations of leptin are commonly observed in this syndrome. 59
Compelling evidence, which implicates POMC in human energy balance, came from a report of two children harbouring loss of function mutations in this gene. 32 The phenotype was characterized by a neonatal history of hypoadrenalism, childhood-onset obesity and hyperphagia, with pale skin and red hair. Rather interestingly, these phenotypic traits can be directly attributable to the absence of an effect of specific POMC-derived peptides on the requisite receptor. Thus, the hypoadrenalism is related to a lack of adrenocorticotrophic hormone (ACTH) action on MC2R, the obesity is the result of loss of α-MSH action on MC4R, and the skin and hair traits are a consequence of diminished α-MSH action on MC1R. Whether the absence of other POMC-derived peptides, such as β- and γ-MSH or β-endorphin, contribute to this phenotype in a subtle way remains to be determined, although recent genetic studies suggest that this may be the case for β-MSH. 60
Further evidence in support of the key role of the melanocortin system in the regulation of body weight in humans came from the identification of mutations in the PC1 gene. 31,61 The phenotype observed in two reported cases, both demonstrating compound heterozygosity for mutations of this gene, included severe childhood-onset obesity due to abnormal processing of POMC. Other features included hypocortisolaemia, hypogonadotrophic hypogonadism, postprandial hypoglycaemia and small intestinal dysfunction, all of which may be related to dysfunctional processing of specific prohormone systems.
As indicated previously, the MC4R appears to function at a critically important juncture in the leptin-mediated pathway effecting mammalian energy homeostasis. The relevance and importance of MC4R in human body weight regulation has also now been confirmed by the identification of several mutations in this gene, which is a highly heterogeneous locus. 30,50,51,62–64 In fact, mutations in the MC4R gene represent the most common monogenic cause of human obesity, with the prevalence of mutations varying between 0.5 and 6.0% of obese subjects. The wide variation in prevalence is, in part, related to selection criteria used in individual studies, with the highest rates being encountered in studies involving subjects with severe early-onset obesity, as opposed to adult-onset disease. Ethnic differences may also play some role in this discrepancy. The characteristic phenotype includes hyperphagia beginning in childhood, accelerated linear growth, increased age-related bone mineral density and hyperinsulinaemia. Extensive phenotype–genotype studies indicate that the degree of hyperphagia can be predicted by the severity of receptor dysfunction caused by individual mutations in in vitro reporter assays. 65 In addition, the concentration of hyperphagia appears to decline with age. 65
While it was initially assumed that the mutations in MC4R were dominantly inherited and had 100% penetrance, it is now apparent that a more complex mode of inheritance is in operation. Thus, although some mutations clearly demonstrate autosomal dominant inheritance, the existence of kindred in which homozygotes for a MC4R mutation manifest more severe obesity than heterozygotes suggest that co-dominance, with modulation of expressivity and penetrance, is a more appropriate method of describing the inheritance pattern. 65 It is likely that other genetic and also environmental factors modulate the phenotypic expression of MC4R.
Leptin resistance in human obesity
In view of its dramatic effect of inducing loss of fat mass in rodents, leptin was, for a brief period after its discovery, hailed as a major breakthrough in the treatment of obesity. However, it was soon realized that circulating leptin correlated with fat mass and BMI and, as a result, that obese individuals had elevated plasma leptin concentrations rather than reduced concentrations. 66 Thus, obesity in humans appeared to represent a form of ‘leptin resistance’ and it was realized that attempts to use exogenous leptin were unlikely to induce significant weight reduction. Indeed, the results of early trials with recombinant leptin bore out these considerations. 67
A number of possible mechanisms have been posited to explain the state of leptin resistance in obesity. 38 These include, impaired leptin transport across the blood–brain barrier and the presence of negative regulators of leptin signalling at the concentration of neurons in the CNS that express LEPRs and their downstream targets. Recently, particular attention has focused on the negative regulatory role of suppressor of cytokine signalling 3 (SOCS3), which inhibits leptin signalling through the JAK/STAT pathway. 68
In relation to the concept of ‘leptin resistance’, it must be borne in mind that leptin most likely plays its intended physiological role at low concentrations and thus, at a biological level, leptin may never have been intended to operate at the exorbitant plasma concentrations seen in obese subjects. The notion that the primary role of leptin is to sense the transition between the starved and adequately nourished state, rather than prevent obesity, is consistent with this idea. 3
Insulin as a lipostatic hormone
Prior to the discovery of leptin, it was proposed that insulin may act as a sensor for energy stores between the periphery and the brain. Certainly, insulin fulfils many of the criteria associated with a lipostatic hormone. 69 In particular, insulin can be transported across the blood–brain barrier in proportion to body fat content, central administration is associated with a reduction in food intake in mammals, and insulin receptors (IRs) are located within the appetite regulatory regions of the hypothalamus. Furthermore, insulin is a key regulator of the switch between fasted and fed states, and plasma concentrations decrease during fasting and increase in obesity.
Further elucidation of insulin's lipostatic role occurred through the creation of mice with neuron-specific disruption of the IR (NIRKO mice). 70 While IR inactivation had no effect on brain or neuronal development, these animals did develop a diet-sensitive obesity associated with increased body fat, mild hyperleptinaemia, insulin resistance and hypertriglyceridaemia. Notably, the level of obesity was mild in comparison with ob/ob mice and also it is evident from leptin-deficient human subjects that insulin cannot substantially compensate for the absence of functioning leptin.
Currently, it is considered that insulin acts in concert with leptin to influence hypothalamic control of energy homeostasis, but the extent and nature of this partnership has to be clarified. In addition, signalling through CNS, IR may modulate the metabolic effects of peripherally acting insulin in the liver and other target tissues. 38
Additional central nervous system pathways
There are an increasing number of other neurotransmitters and neuropeptides, and their associated receptors, for which a specific role in the control of food intake and overall energy balance has been proposed. 48 In humans, serotonin (5-HT) is a monoamine with many diverse CNS effects, including the process of satiation, and these effects are mediated through at least 14 subtypes of receptor. Brain-derived neurotrophic factor and its receptor TrkB have also been implicated in the central regulation of energy balance. Of note, a recent report described a child with a de novo mutation in NTRK2, the gene that encodes the neurotrophin receptor, TrkB. This mutation markedly impaired receptor autophosphorylation and downstream signalling, resulting in hyperphagic obesity and learning disability. 71
Other neuropeptides and receptors with potential central effects on feeding include, CB1 cannabinoid receptor (CNR1) and the endocannabinoids, anandamide and 2-arachidonoyl glycerol, interleukin-6 (IL-6), neuromedin U, ciliary neurotrophic factor (CNTF), cortocotrophin releasing factor (CRF), urocortin and orexins. 48
Gut hormones in the regulation of energy balance
As indicated previously, leptin and insulin provide a mechanism by which adipose tissue can signal the hypothalamus and influence overall energy balance (Figure 2). However, it is well recognized that the control of appetite is based on a highly complex network of interactions and that signals from the gastrointestinal tract can also impact on appetite control and energy homeostasis. 72 In particular, it has been hypothesized that gut hormones play a significant role in postprandial satiety. 73
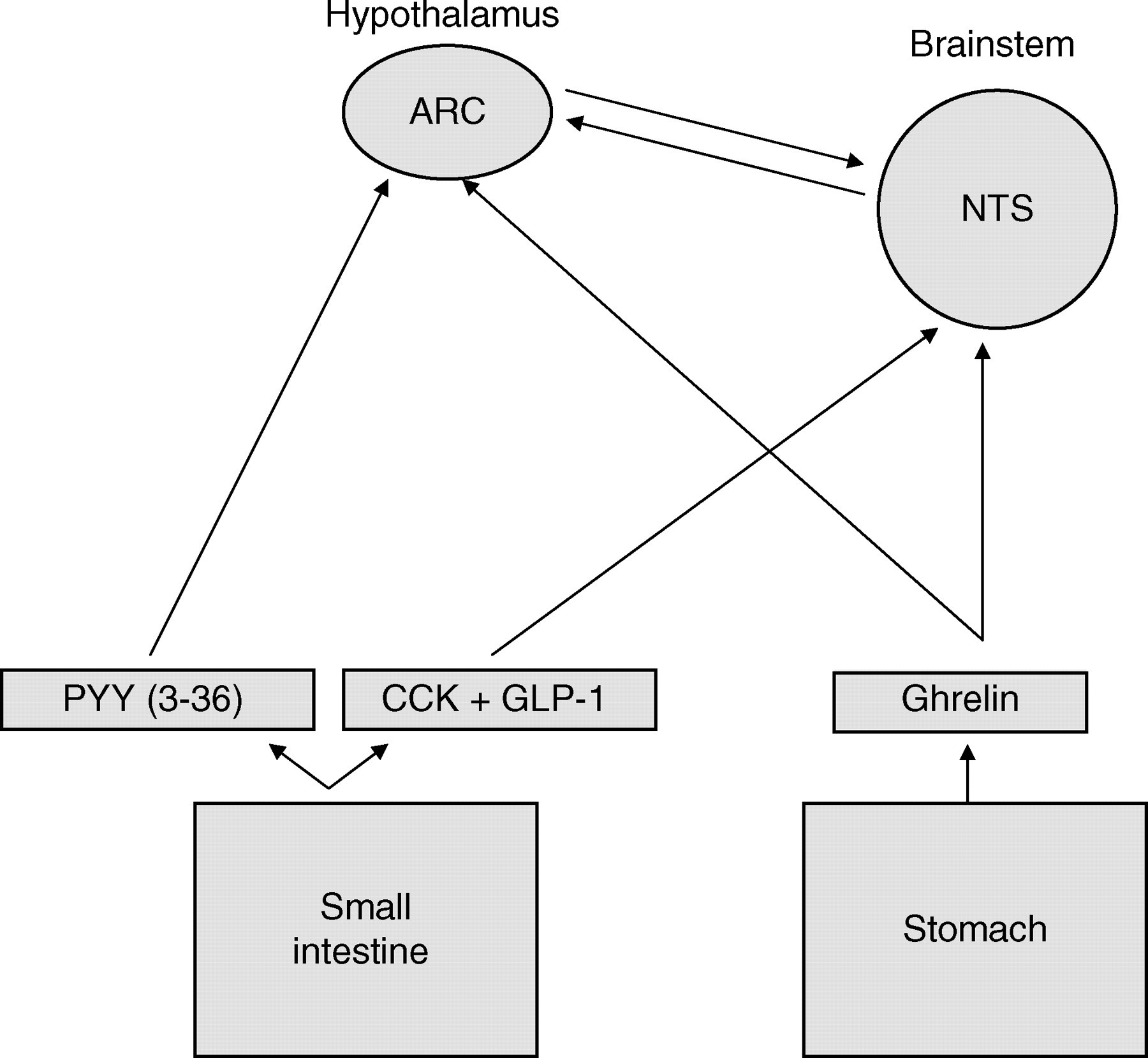
Sites of action of gut-derived signals influencing food intake. ARC, arcuate nucleus; CCK, cholecystokinin; GLP-1, glucagon-like peptide-1; NTS, solitary nucleus; PYY3-36, peptide YY3-36
There is an abundance of research supporting the role of CCK in mediating meal termination. 74,75 In essence, the presence of luminal fat and protein stimulate the release of CCK from duodenal mucosal cells. In turn, CCK activates CCKA-type receptors located on peripheral vagal afferent fibres, which transmit signals to the NTS in the brainstem and the signal is relayed from this site to the hypothalamic feeding centre. 76,77 Of note, the effect of infused CCK is relatively short-lived (30 min), which is consistent with its role as a mediator of satiety and meal termination. 77
GLP-1 is also implicated in appetite regulation, and is derived from the cleavage of the precursor preproglucagon in pancreas, intestinal L cells and the CNS. 78 It is released from the gut into the bloodstream in response to intestinal nutrients and GLP-1 receptors are found in the brainstem and hypothalamus (PVN and ARC). 75,77 ICV administration of GLP-1 suppresses appetite, 79 while ICV injection of a GLP-1 antagonist increases food intake. 80 GLP-1 is also recognized as an incretin, potentiating insulin secretion from the pancreatic islets, and this may contribute to its role in satiety. 81 Overall, the biological importance of GLP-1 as both incretin and neuropeptide remains controversial and requires further elucidation.
Peptide YY (PYY) is co-secreted with GLP-1 from intestinal L cells, primarily as the PYY3-36 form, in response to gut nutrients. 75,82 It is related to and has approximately 70% amino acid homology with neuropeptide (NPY). However, recent studies involving both rodent and human subjects, have reported that peripheral administration of PYY3-36 leads to a marked inhibition of food intake in contrast to the effects of NPY. 83–85 Notably, these effects are not observed in transgenic mice lacking the NPY Y2 receptors, implicating this receptor as the mediator of the PYY3-36-related anorectic action. 83 Of note also is that natural plasma concentrations of PYY3-36 were lower in obese subjects compared with lean controls. These findings have generated substantial interest in this peptide as a potential treatment for obesity. 86
A number of other gut and pancreatic peptides released postprandially, which may have specific effects on inhibition of food intake, include gastric inhibitory peptide (GIP), amylin, enterostatin and ApoA-IV. Oxyntomodulin (OXM) is another product of preproglucagon post-translational processing and it has very similar anorectic effects as GLP-1 in both rodents and humans. 87,88 Intravenous infusion of OXM has been noted to reduce plasma ghrelin concentrations in humans. 89 Further work is required to determine the precise role of OXM in human appetite regulation and its potential as a treatment option for obesity.
Ghrelin is another gut-derived hormone that has generated a substantial level of interest recently. This peptide is synthesized in the stomach and is the endogenous ligand for the previously identified orphan receptor, growth hormone secretagogue receptor (GHS-R), so called because activation of GHS-R in the hypothalamus resulted in pituitary growth hormone release. 90 Ghrelin contrasts with other gut hormones in that its primary action involves stimulation of food intake, and indeed, chronic administration in rodents leads to obesity. 91 It is also a potent stimulator of food ingestion in humans on peripheral administration. 92 Ghrelin concentrations are higher during fasting state and concentrations rise further just before spontaneous feeding, strongly suggesting a role in meal initiation. 93 Plasma concentrations of ghrelin are low in obesity and interestingly a fall in ghrelin concentrations after gastric bypass surgery, with an associated loss of pre-meal peak, has been posited as a factor contributing to the suppression of appetite observed with this procedure. 87,94
Integrated mechanisms regulating energy intake
With the recent advances in our understanding of the complex pathways involved in regulating appetite, satiety and body nutrient stores, an integrated model of these processes can be constructed (Figure 3). In essence, it would appear that the short-term regulation of food intake is determined, in part, by both neural and endocrine meal initiation and meal termination signals from the gastrointestinal tract. In addition, circulating concentrations of blood metabolites, e.g. glucose, amino acids and fatty acids, also contribute to these regulatory mechanisms. In relation to long-term body ‘energy stores’, lipostatic signals such as leptin and insulin mediate between the peripheral adipose tissue and the central neuroendocrine pathways in the hypothalamus and beyond to maintain energy homeostasis. This represents a simplistic model and undoubtedly, in the complex energy homeostatic milieu, gut-derived appetite regulating hormones and the ‘lipostatic’ hormones have effects across the spectrum of both short- and long-term energy balance. Moreover, there is no doubt that higher brain centres controlling complex behaviour patterns provide a major input into both short- and long-term pathways, modulating these responses.
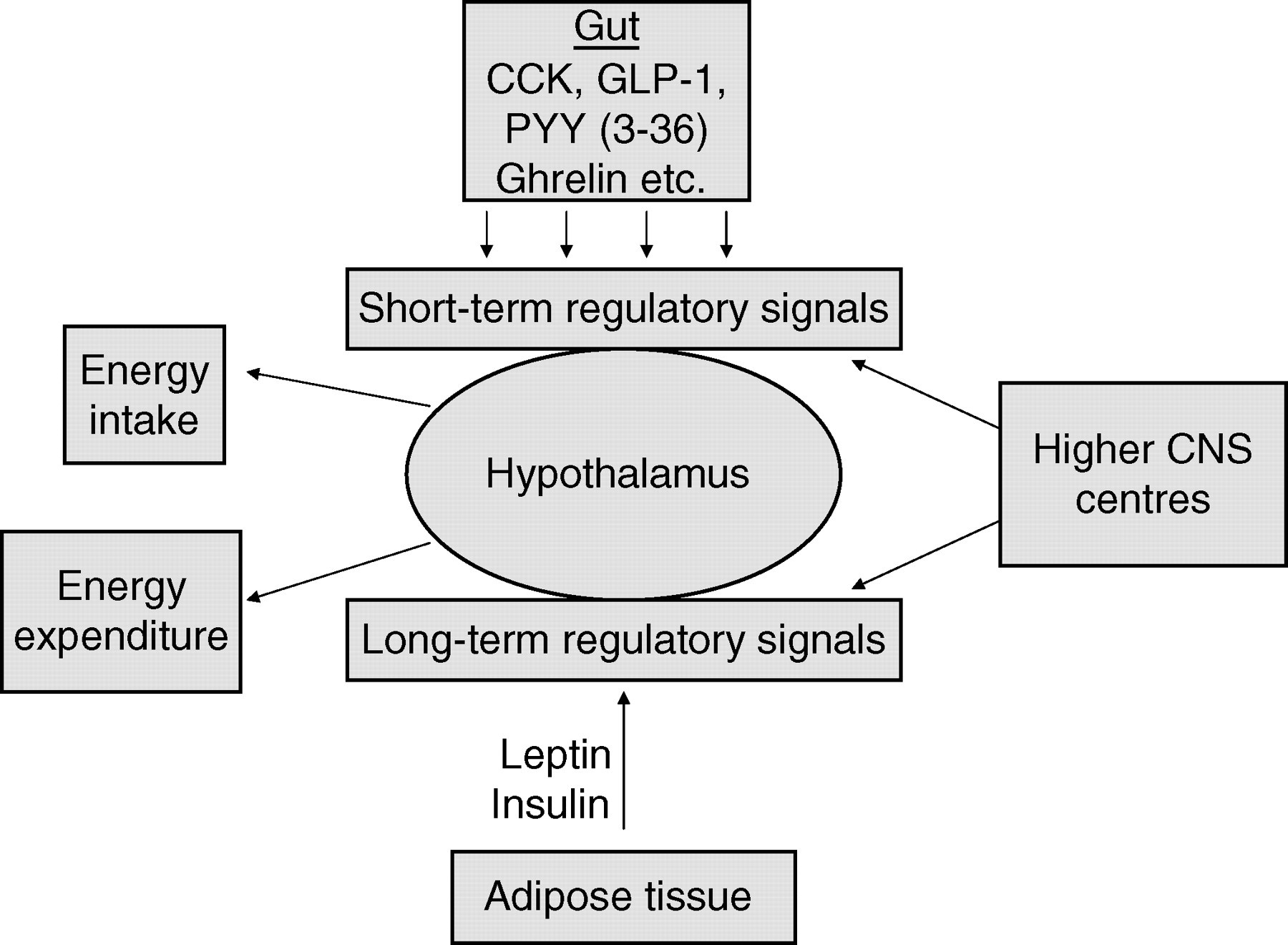
Integrated model of short- and long-term energy intake control CCK, cholecystokinin; GLP-1, glucagon-like peptide-1; PYY3-36, peptide YY3-36; CNS, central nervous system
Management of obesity
Clinical approach to the obese patient
While the major objectives of public health strategies are aimed at prevention of obesity, it is critically important that detection and management of established obesity and the associated co-morbidities are emphasized in current clinical practice. 6,7,19,95,96 The problem of obesity has reached such a level that health-care institutions in the UK and Ireland are now recognizing the need for specialized obesity clinics to tackle many of these issues. In fact, any effective clinical strategy will require a multidisciplinary approach with input from medical staff, including general practitioners and hospital-based specialists, dieticians, clinical psychologists and physiotherapists among others. 96
In relation to the medical approach to obese subjects, a clear medical history including assessment of co-morbidities, e.g. diabetes, CVD and operative history, should be ascertained. In view of the fact that many common drugs, e.g. antidepressants, insulin, beta-blockers, corticosteroids, anticonvulsants and the oral contraceptive pill, can cause or exacerbate weight gain, a lucid medication history is also important. The overall weight and dieting history is essential as is an assessment of physical activity. Obesity is often familial, either as a result of shared environment, genetics or the combinations of these two factors. In addition, many obese subjects suffer with low self-esteem and there may be clear psychological issues to be addressed as either a cause of morbidity or a barrier to successful treatment. In female patients, issues relating to fertility should be broached.
Any examination in the context of obesity must include a minimum set of anthropometric measures of obesity, including BMI, WC and blood pressure. Baseline investigations should encompass a biochemical profile, with ‘renal profile’, ‘liver profile’, ‘thyroid function tests’, as well as fasting lipid profile and glucose. In suspected Cushing's syndrome appropriate plasma and urine screening investigations should be performed. In female patients with features suggestive of polycystic ovarian syndrome (PCOS), concentrations of gondotrophins, relevant sex hormones and sex hormone-binding globulin (SHBG) should be measured. Of note, low concentrations of SHBG are encountered in PCOS, but suppression of SHBG is also a feature in male subjects with morbid obesity. In addition, both total and free testosterone concentrations are low, although gonadotrophins are usually normal, indicating that obesity-related biochemical hypogonadism is most probably clinically insignificant. 97 Of course, caution needs to be exercised in interpreting such data in the presence of hypothalamic or pituitary pathology.
The presence of co-morbidities or aberrant results of baseline tests may precipitate further investigations such as an oral glucose tolerance test (OGTT), haemoglobin A1c (HbA1c), echocardiogram or exercise stress test, radiological imaging. While obesity may be associated with normal fasting glucose concentrations or an OGTT, underlying insulin resistance may still be present. Thus, measurement of indices such as fasting glucose to insulin (G/I) ratio may have a role to play in documenting the degree of insulin resistance, particularly in paediatric obesity and PCOS. Caveats relating to cross-reactivity with proinsulin and the absence of standardized insulin assays indicate that caution should be exercised in the interpretation of the G/I ratio, particularly between laboratories.
Elements within the clinical history and examination such as severe early-onset hyperphagia or obesity, a strong family history of obesity, associated developmental delay and the presence of dysmorphic features should suggest the possible need for molecular genetic and cytogenetic analysis. In children with a suggestive history, and in whom obesity is the predominant or only disorder, measurement of serum leptin and plasma insulin and proinsulin may give insight into the presence of congenital leptin or PC1-deficient states, respectively. 98 It should be noted however that contrary to what was initially reported, markedly elevated concentrations of serum leptin are not a feature of inherited LEPR deficiency. 59 Detection of MC4R-deficient state is primarily dependent on mutation scanning of the MC4R gene. 98
Treatment options in obesity
In planning treatment programmes for obesity, it is important to bear some fundamental issues in mind. Obesity is a chronic problem and therefore will most likely require life-long management, as maintenance of weight loss can be very difficult to sustain. Any treatment targets should be realistic and agreed with the patient concerned. Moreover, different patients may require different approaches, as no single treatment is effective in everyone. In terms of outcome, a 10% reduction in baseline weight is likely to substantially benefit patients in terms of metabolic morbidity.
The cornerstone of all obesity treatment programmes is lifestyle modification with specific emphasis on diet and physical activity levels. 6,7,95,96 Dietary restrictions can range from the more traditional low or very low calorie diets through to high-protein–low-carbohydrate diets, such as the Atkins diets or the low-glycaemic index (GI) diets, both of which are currently receiving a high media profile, although medical opinion on their value remains to be defined. Exercise and dietary regimens may be combined with specific behavioural therapy measures.
Pharmacotherapeutic intervention is usually considered if a 5% reduction in weight is not attained within a minimum of three months of lifestyle modification. 96,99 While many drugs may have an anti-obesity action, currently National Insitute for Health and Clinical Excellence (NICE) guidelines recommend the use of either orlistat or sibutramine in the treatment of obesity in the UK. Orlistat is an intestinal lipase inhibitor and effectively induces a partial state of fat malabsorption, with 33% of dietary fat remaining unabsorbed. It is unsurprising, therefore, that the principal side-effects of this medication relate to GI disturbances, including bloating, flatulence, diarrhoea and abdominal pain. 96,99 Sibutramine acts in the CNS and inhibits re-uptake of both serotonin and noradrenaline. Thus, its effects include an anorectic effect through activation of 5-HT receptors, including 5-HT2c, and α- and β-adrenergic receptors within the CNS. It may also promote thermogenesis, although this probably plays a minor role in its weight-reducing action. Sibutramine has a moderate side-effect profile with problems such as dry mouth, headache, insomnia and constipation being encountered. One particular issue relates to a possible increase in both systolic and diastolic blood pressure, thus requiring careful follow-up in treated patients. Both orlistat and sibutramine are reported to induce a 5–10% weight loss from baseline, and are particularly effective if combined with adequate lifestyle measures. These agents are recommended for use in patients with BMI > 30 kg/m2 or BMI > 27 kg/m2 in the presence of co-morbidities and should be used under close medical supervision. 96,99
In recent years, evidence has emerged implicating the endocannabinoids, anandamide and 2-arachidonoyl glycerol, and their associated receptor CB1 in the regulation of food intake. More specifically, it has been noted that CB1 receptor gene (Cnr 1) knockout mice eat less than wild-type littermates. In addition, defective leptin signalling on ob/ob and db/db mice is associated with elevated hypothalamic concentrations of endocannabinoids. This has inevitably led to the development of selective CB1 anatagonists by a number of pharmaceutical companies. Rimonabant is currently the only CB1 receptor with European agency for the Evaluation of Medicinal Products (EMEA) approval for clinical use. 99 Based on double-blind placebo-controlled randomized multicentre trials, i.e. rimonabant in obesity (RIO)-North America, RIO-Europe, RIO-Lipids and RIO-Diabetes, it would appear that a dose of 20 mg of rimonabant, in combination with a hypocaloric diet, can lead to significant (>5%) weight reduction. 100–102 Moreover, observed improvements in HBA1c, dyslipidaemia and inflammatory markers suggest that rimonabant may address not only obesity per se, but also major co-morbidities, and thus could lead to a reduction in the number of medications needed to treat obesity and its associated cardiometabolic risk. 102 Adverse effects have been reported including nausea, dizziness, diarrhoea and insomnia. Psychiatric disorders, mainly depression, were reported in 6–7% of rimonabant-treated individuals, warranting discontinuation. 99 Therefore, this class of drug needs to be used with caution in the presence of significant psychiatric morbidity and may well be contraindicated. However, further studies are ongoing to determine the exact extent of its use in clinical practice and, of note, the Food and Drug Administration has held back approval in the USA pending the outcome of these studies.
Surgical treatments of obesity are usually reserved for morbid obesity (BMI > 40 kg/m2). Various bariatric procedures are used in practice, including laparoscopic gastric banding and gastric bypass. Surgical treatment can be very effective and may substantially reduce mortality associated with morbid obesity. 96
Obesity epidemic: implications for clinical biochemistry
The spiralling increase in the prevalence of obesity and allied health problems such as, T2DM and CVD, has led to the realization that the management of obesity is now an intrinsic component of mainstream medical practice. However, despite the huge number of patients afflicted with this condition there is still a long way to go in terms of providing a comprehensive clinical service dedicated to managing obesity in many hospitals. Obesity is fundamentally a metabolic disorder and as such chemical pathologists and metabolic physicians are well situated to address this service deficit. Indeed, exposure to the management of obesity and its co-morbidities should be a core aspect of training in metabolic medicine and chemical pathology.
Clearly, the provision of a substantive clinical obesity service requires a comprehensive clinical biochemistry laboratory. In particular, the investigation of the causes of obesity requires a repertoire of general biochemistry and endocrine tests. Of course, this also has implications for workload within such laboratories in view of the high prevalence and continuing epidemiological trends of obesity.
The elucidation of the molecular mechanisms underlying appetite control and satiety brings with it the prospect of developing new diagnostic assays for use in the investigation of obese patients. Even at this stage one could cogently argue that the measurement of plasma leptin should be available in centres with specialist obesity clinics, particularly involving paediatric practice. Similar arguments could be made in favour of ghrelin and PYY3-36. Moreover, the prevalence of MC4R mutations in childhood-onset severe obesity is such that the inclusion of MC4R molecular diagnostics in regional biochemical genetics laboratories is worth considering. As should be the case for any specialized laboratory test, protocols for use and interpretation of such tests will require substantial input from clinical biochemistry personnel.
Another factor to consider is the role of clinical biochemistry laboratory in both proactively promoting research into the realm of obesity and facilitating such research. Certainly, there are already a number of major clinical biochemistry laboratories in the UK involved in internationally recognized research in this area and this may expand in the future.
Conclusions
Obesity is a major public health issue. Understanding the physiological and molecular mechanisms regulating energy balance will inform the development of effective prevention, diagnostic and treatment strategies for this condition, and over the last decade enormous progress has been made in this field. Undoubtedly, clinical biochemistry has a role to play in facilitating such initiatives.
Footnotes
Acknowledgements
Many thanks to Professor John Nolan, Dr Sadaf Farooqi, Dr Giles Yeo, and Professor Steve O'Rahilly, with whom discussion of various issues covered in this manuscript was very helpful in its preparation.