
Abstract
Abstract
Screening newborn babies for inherited metabolic disease began in the UK in the late 1950s with the ‘nappy test’ for phenylketonuria. In 1969 the Department of Health recommended changing to bloodspot screening using the techniques developed in the USA by Robert Guthrie and his associates. Bloodspot screening for various other disorders (galactosaemia, maple syrup urine disease, homocystinuria, cystic fibrosis and others) was introduced on a patchy local basis but, until 2000, the only additional disorder officially recommended was congenital hypothyroidism. Screening for haemoglobinopathies received official support in 2000 and for cystic fibrosis in 2001 though implementation was slow, particularly for the latter. Both these screens have raised difficult issues relating to genetic privacy and the detection of carrier status in children. During the last decade screening has become increasingly subject to central control. Though a more consistent and systematic approach was clearly needed, this has undoubtedly slowed the rate of innovation. In particular the UK has lagged behind many other European countries in the application of tandem mass spectrometry (MS-MS) though, following a major pilot study, screening for medium-chain acyl-CoA dehydrogenase deficiency is now in the process of introduction. Attempts to codify clinical and laboratory procedures have also proved controversial, highlighting marked differences in practice in various parts of the country and the difficulty of rationalizing these within a practicable and scientifically justified framework. Notwithstanding this, there are many positive developments and newborn screening remains a stimulating and rewarding field in which to work.
The past
‘
Newborn screening for metabolic disease in the UK began in the late 1950s with small, locally-organized programmes for the early detection of phenylketonuria. Prompt dietary treatment soon after birth had been largely successful at preventing the otherwise severe mental retardation associated with this condition and a suitable urine test was available in the form of ferric chloride solution (or later the Phenestix® test strip) applied to the wet nappy. The test was performed at 4–6 weeks of age by Health Visitors, at that time employed by local authority health departments. In 1960, a conference organized by the Medical Research Council recommended that ‘local authorities should continue to maintain and, if possible, to expand their present programme of routine screening tests’ and by May 1962 136 of the 145 local authorities in England and Wales were either actually screening or were planning such schemes. 2 The technically superior Guthrie test, using dried blood on filter paper and a bacterial inhibition assay, was introduced in Scotland in 1964, covering the whole of that country by 1968. In 1969, after some four years of deliberation, the Department of Health (England and Wales) recommended switching to the blood-based screen. Responsibility was split. Collection of the blood sample and checking that all babies had been tested was to be organized at local authority level, overseen by the Medical Officers of Health. Regional Hospital Boards were responsible for setting up regional or supra-regional laboratory services, to be closely associated with diagnostic and treatment centres. The modern era of newborn metabolic screening had begun.
There was a considerable scope for local experiment and diversity in the early years, despite a degree of central oversight through the UK Phenylketonuria Register and its Steering Committee. 3 Several screening centres replaced the Guthrie bacterial inhibition assay for phenylalanine by paper or thin layer chromatography or more quantitative methods, mainly continuous flow fluorimetry. The former could also detect, among other disorders, maple syrup urine disease and pyridoxine non-responsive homocystinuria 4 while the more sensitive quantitative methods allowed the majority of cases of galactosaemia to be identified by a secondary increase in blood phenylalanine concentration. 5 However, for the first 30 years following the introduction of bloodspot screening the only additional test to be officially recommended throughout the UK was that for congenital hypothyroidism. 6
Screening for congenital hypothyroidism started in Quebec in 1974. Radioimmunoassay was still in its infancy and screening had to be based on thyroxine. 7 Affected babies often had thyroxine concentrations within the reference range and it was necessary to have a second-tier test for samples with results below the 20th centile. The second tier was thyrotropin in liquid blood or later, as techniques improved, in the dried blood sample. This two-stage method detects both primary hypothyroidism, due to agenesis or hypoplasia of the thyroid gland or to defects in thyroxine biosynthesis, and secondary hypothyroidism due to pituitary dysfunction. However, secondary hypothyroidism proved to be quite rare and, in line with most other European countries, the UK adopted a single-stage strategy based on thyrotropin alone. 6,8 East Anglia, which had pioneered screening for hypothyroidism some years earlier, retained the two-tier thyroxine-thyrotropin method for several years.
The official directive introducing screening for hypothyroidism, HN(81)20, 6 was published some four years after the Joint Standing Sub-Committee on Screening in Medical Care first recommended setting up a trial service. Its appendix makes interesting reading, describing how passage through three further committees lead eventually to the realization that a trial was no longer ethical or possible as much of the country had already started a routine service. The arguments had been well researched; the case for screening was made very systematically and included a cost-effectiveness analysis. Thyrotropin concentrations >80 mU/L whole blood were regarded as ‘positive’, results between 25 and 80 being classed as ‘suspicious’. These values reflected the performance of the commercially available kits at the time – the assay took 48 h to complete. Currently the UK Newborn Screening Programme Centre 9 recommends that babies with initial results between 10 and 20 mU/L should be considered as borderline and some screening laboratories are using even lower cut-off values. Some of these changes can be attributed to differences in assay calibration but ‘technology drift’, where action levels are determined by what is analytically achievable irrespective of clinical significance, is an ongoing issue. The situation is complicated in that in some affected infants, particularly those born prematurely or with thyroxine synthesis defects, hyperthyrotropinaemia may be delayed. Marginally increased thyrotropin in the initial screening sample may portend overt hypothyroidism though in the majority of cases it does not and for these the need for medical intervention has yet to be convincingly demonstrated. The difficulty is to balance the wish to maximize screening sensitivity against the need to minimize the number of repeat blood samples requested and unnecessary follow-up investigations. The value of routinely collecting a second blood sample from all babies born prior to 35 weeks gestation is currently under investigation in the UK.
Newborn screening often proved controversial. Surprising as it may now seem, even screening for phenylketonuria was not accepted without heated debate, particular in the USA where it is mandatory in many states. 10 There were claims that the benefit of early treatment could be due to placebo effects and in 1968 the UK Medical Research Council considered a randomized-controlled trial. Fortunately this was rejected on the advice of concerned paediatricians. The broader debate continued, however, against a background of general interest in expanding screening of newborns and for a range of adult diseases such as breast and cervical cancers, diabetes mellitus and glaucoma. However, it was becoming evident that many screening programmes provided only limited benefits and most had adverse effects. By 1980 Guthrie 11 was able to list over 20 bloodspot screening tests for inherited metabolic disease, but few passed into routine use. There were several attempts to produce universal criteria against which screening programmes could be evaluated. The best known are the 10 ‘principles’ of Wilson and Jungner 12 (Table 1) which have been widely accepted though they are not entirely appropriate for newborn screening as they largely ignore the family dimension. 13,14
Principles of early disease detection 12
When compared with many Western countries, UK newborn screening policy was very conservative 15 though the regional structure of the National Health Service (NHS) allowed limited local developments. Scotland was particularly active and until recently screened specifically for galactosaemia. Screening for cystic fibrosis using dried bloodspot immunoreactive trypsinogen was introduced to the UK in East Anglia in 1980 and by 1997 had been extended to an additional six areas, including Northern Ireland and Wales (see below). Bloodspot screening for sickle cell disease and other haemoglobinopathies started in the City of Birmingham in 1979. In Wales, screening for Duchenne muscular dystrophy started in 1990 and has been well received. Even though no recognized treatment is available, early diagnosis can ease the family journey and knowledge of the disease may influence future reproductive choices. 16
The present
In recent years, we have moved from an NHS with a degree of devolved responsibility for screening matters to one where screening is tightly regulated centrally through a UK-wide National Screening Committee (NSC) formed in 1996. 17 The two reports from this Committee provide an excellent insight into the underlying philosophies of screening and the issues that need to be considered when a candidate programme is being assessed. There can be no doubt as to the quality and the quantity of the intellectual input. The original 10 principles of Wilson and Jungner have been expanded to 19 criteria (Table 2), with a ‘Format’ for the extensive set of data to be collected for the assessment. While it is recognized that not all the criteria and questions raised in the Format will be applicable to every proposed programme the degree of rigour generally required by the NSC can be judged from Criterion 11: ‘There must be evidence from high quality randomized-controlled trials that the screening programme (unless aimed solely at providing information to allow the person being screened to make an informed choice) is effective in reducing mortality or morbidity’.
National Screening Committee Second Report: Criteria to be met before screening for a condition is initiated17
The UK Health Technology Assessment Programme also contributed to the development of screening policy, commissioning two reports on newborn metabolic screening. 13,18 Both of these reports considered the potential of ‘expanded screening’ for a range of disorders using mass spectrometry (MS-MS) to detect increased concentrations of amino acids or acylcarnitines. Both felt that this new technique had great potential, but needed detailed evaluation before passing into routine service. To this end both recommended pilot or research studies, special emphasis being given to medium-chain acyl-CoA dehydrogenase (MCAD) deficiency as being a relatively common disorder and fairly easily managed. Later Health Technology Assessment reports dealt with the newborn screening for cystic fibrosis and for haemoglobinopathies. 19 These three fronts have progressed at very different speeds.
Screening for haemoglobinopathies received a high priority. The NHS Plan (2000) promised linked antenatal and neonatal screening programmes for ‘haemoglobinopathy and sickle cell disease’. The newborn component has been introduced in England, helped by pump-priming funding from the Department of Health, with national coverage achieved in July 2006 making England the first country in Western Europe to introduce such a universal screening programme. It is based on existing screening laboratories though the requirement for a minimum throughput of 25,000 samples per year has led to closure of several of the smaller units. Antenatal screening has progressed more slowly and will not be centralized to the same degree. In contrast to neonatal screening, which is universal, in low-prevalence areas antenatal screening will be offered only to high-risk ethnic groups, full implementation is expected by mid 2007 (NHS Sickle Cell and Thalassaemia Screening Programme;
Newborn screening for haemoglobinopathies is targeted primarily at sickle cell diseases (HbSS, HbSC, HbSD and HbSE) where there are demonstrable benefits from early prophylactic treatment. Severe forms of thalassaemia may also be detected. Screening is either by high-performance liquid chromatography (HPLC) with isoelectric focusing as a follow-up test, or vice versa. Both techniques provide a full haemoglobin profile and detect carrier states and a variety of rarer haemoglobin variants. In the UK, it is now regarded as mandatory to report carrier status for clinically significant haemoglobinopathies, though this has not always been the practice and there has been much debate on the ethics of disclosing carrier status in children. 20 There is also controversy over whether haemoglobin variants with no known clinical significance should be reported, an example of the clash between the basic principles of screening on the one hand and practices that have grown up in a completely different clinical setting on the other. Babies who have received a blood transfusion before the screening sample is taken need to be retested four months later and this, together with the counselling of families of carrier babies, has proved to be an unexpectedly heavy burden on health resources. Even in medium-prevalence areas approximately 1% of all babies screened require some form of follow-up action. Furthermore, many of the babies shown on newborn screening to be affected with sickle cell diseases transpire to have been diagnosed locally before the screening result is available.
Screening for cystic fibrosis is being expanded as a result of pressure exerted by a parent-led support group. 21 The NSC's view was that benefits of newborn screening have yet to be convincingly demonstrated (though information continues to accumulate 22 ) but as 20% of the UK population was already being screened, expansion to cover the remainder of the country could be justified on the grounds of equity. However, here the official announcement in April 2001 was followed in England by a four-year hiatus before limited pump-priming funding became available (Northern Ireland had been screening for cystic fibrosis since 1983 and Wales since 1997. Scotland started screening in February 2003). The intention is to bring all remaining areas of England into the programme by April 2008. The screening protocol for cystic fibrosis 23 is quite complex, involving both bloodspot immunoreactive trypsinogen assay and mutation analysis, and seeks to balance sensitivity, specificity and regional and ethnic differences in cystic fibrosis transmembrane conductance regulator (CFTR) genotypes. The inclusion of a mutation analysis step has proved controversial as the screen will identify a small group of ‘low-risk’ babies, almost all unaffected carriers, whose families need to be counselled in a careful and balanced manner. The protocol has been designed to minimize the number of families involved: only 0.5% of screened babies are genotyped so that, in contrast to the sickle cell screen, only a small proportion of carrier babies will be thus identified.
Mass screening by MS-MS has only been really practicable since 1998, but the technique has been taken up rapidly in many countries worldwide. It can replace conventional methods of screening for phenylketonuria and can detect a wide range of other disorders of intermediary metabolism, most of which are well known to diagnostic laboratories but which, with a few exceptions, have never been the target of mass screening. For reasons that are perhaps understandable 14 the two 1997 health technology assessment reports 13,18 were greeted initially by considerable scepticism and it was not until March 2004 that a multicentre collaborative pilot study limited to MCAD deficiency was launched. Over a period of two years data from screening around 700,000 UK newborns (6 screening laboratories covering approximately half of UK births) were collected, the unscreened areas acting as controls. The study included technical performance of the screen itself, diagnostic and clinical management protocols, assessment of clinical outcomes up to the age of two years, a survey of the psychological impact of screening on the parents, and cost–benefit analysis. Perhaps, because of the relatively late age of blood sampling (day 5 of life is recommended) a simple protocol has proved adequate, with octanoylcarnitine >0.5 μmol/L whole blood giving a positive predictive value of >80%. 24 MCAD deficiency is particularly prevalent in the UK (greater than 1 in 10,000 in the areas screened), but otherwise the results are broadly in-line with international experience.
MS-MS screening requires a clear understanding of the biochemistry and differential diagnosis of the disorders concerned and still presents significant analytical challenges. In the UK pilot study these challenges have been tackled by an in-depth clinician-laboratory collaboration and by a multipronged EQA scheme, with data from the participating laboratories shared in an open, non-anonymized, manner. Screening continues in the pilot regions and the NSC reported favourably on data presented at an interim review in July 2006. After a period of some uncertainty, the Department of Health announced on 7 February 2007 that screening for MCAD deficiency is to be rolled out throughout England over the next two years. ‘The final report (of the study) will be available in 2008, but sufficient evidence and analysis was available for the NSC to make its recommendation that newborn screening of all babies would be clinically and cost effective in the UK.’
The future
There are several possible additions to the UK newborn screening programme: Duchenne muscular dystrophy is screened for in Wales and is generally well accepted 16 and screens for biotinidase deficiency, galactosaemia, congenital adrenal hypoplasia are widely practised abroad, increasingly so in recent years. None of these has met with approval from the NSC. Disappointing, also, has been the lack of progress in applying MS-MS screening to other disorders resulting in abnormal bloodspot amino acid or acylcarnitine profiles. The development of second-line tests, such as succinylacetone as a marker for tyrosinaemia type 1, makes it more practicable to screen these disorders despite the low specificity of the initial test. 25
Internationally, the introduction of ‘extended’ MS-MS screening is proceeding apace, albeit with some startling differences in both underlying philosophy and practice. 26 Screening for some of the lysosomal disorders is being actively explored. 27 In the UK there has been a further review 28 from the Health Technology Assessment Programme, but little serious consideration at the policy level or progress towards practical exploration. The majority of UK screening laboratories now use MS-MS to screen for phenylketonuria, and reagents for the Guthrie bacterial inhibition assay of phenylalanine have been discontinued by many former suppliers. In switching to MS-MS most of the areas previously screening by paper or thin layer chromatography have concurrently restricted the range of amino acid disorders covered (omitting homocystinuria, maple syrup urine disease and tyrosinaemia for example) even though for some there is very clear evidence of the clinical benefit of screening. 29 The NSC's current policy guidance 30 is that screening for these disorders should not be offered.
Screening for phenylketonuria, congenital hypothyroidism, sickle cell disorders and cystic fibrosis had all been introduced as local schemes long before they were adopted as national policy. Experience with these schemes informed such policy and greatly eased the process of expansion. Other local projects, urine screening for neuroblastoma for example, failed to provide the expected benefits and were quietly dropped. While a centralized national approach, with its higher political profile, may have advantages for some aspects of health care, in newborn screening it has ‘raised the stakes’ and tended to stifle innovation. The NSC's published requirements (Table 2) would effectively rule out screening for all but the most common disorders. 31 Even for MCAD deficiency, a relatively common and straightforward condition, a randomized-controlled trial would be protracted and prohibitively expensive 32 and eventually the NSC agreed to a less rigorous study (above). This compromise design would still be impractical for most of the other potential MS-MS screens due to the rarity of the disorders concerned. The impossibility of collecting enough data for a quantitative policy evaluation is insufficiently recognized. 31 Pandor et al. 28 having reviewed MS-MS screening for a wide range of conditions in great detail, reported that ‘it is difficult to draw firm conclusions on extending the UK screening programme to all disorders detectable by MS. … Robust evidence on the underlying incidence and outcomes of many of the disorders was lacking, particularly differences in long-term outcomes that are attributable to therapies initiated as a consequence of presymptomatic detection using tandem MS’ and recommend research to ‘establish the sensitivity and specificity of neonatal screening using tandem MS for other individual inborn errors of metabolism in the UK and to determine the underlying incidence of these conditions. … to ascertain the natural history of some conditions, and the potential economic impact of screening’. Apart from the problem of scale, there is also a degree of unreality in the categoric thinking implicit in a disease by disease approach. Nearly all inborn errors are heterogeneous and display a range of severity and outcomes. Incidence varies greatly with ethnicity and geographic origin. The effectiveness of treatment is continually improving, thus vitiating the use of long-term data. Even where quantitative data are available they are seldom sufficient in themselves and must be converted into a qualitative judgement. For example, the tipping point which determines whether a screening test is ‘precise’ (NSC Criterion 4 [Table 2]) remains undefined. Whether the test is acceptable for its purpose is a subjective, not an objective decision. Ultimately such decisions will have to be made on holistic and pragmatic bases bearing in mind that, after a while, taking no decision becomes a decision in itself. 26
The other area of faltering progress is overall organization. Newborn bloodspot screening involves a wide variety of health professionals working in a range of NHS (and, increasingly, other) organizations. The 1969 Health Department circular introducing bloodspot screening for phenylketonuria allocated responsibilities very clearly, with local authority Medical Officers of Health ensuring that every baby was tested and the Regional Hospital Boards designating laboratory and clinical services. This clarity has not survived the numerous NHS reorganizations of ensuing years and a national audit in 1997 found deficits in almost all aspects of both structure and performance. Local arrangements for ensuring coverage were particularly chaotic. The audit report 33 recommended that urgent priority should be given to … . clarifying national policy for the programme and establishing a formal quality assurance scheme, recommendations echoed in a later health technology assessment report. 13 Eventually, the UK Newborn Screening Programme Centre was established and published its first set of policies, standards and guidelines in April 2005. 9 These incorporated the output of several ad hoc multidisciplinary working groups on a range of topics (information and consent, blood sampling, standards relating to coverage and timeliness, interpretation and clinical referral, information systems and registers) and brought together many strands of current practice. However, various aspects, ranging from how the blood sample should be taken to what cut-off levels should be set, have proved controversial. Local practices relating to screening cut-offs, repeat sampling, and subsequent clinical management vary considerably and a rigorous scientific review is required. On the audit front, the first round of data collection revealed continuing problems in ensuring complete coverage. The resultant concern at Strategic Health Authority level is being overshadowed by another round of reorganization. Equally disappointing is the lack of progress on a screening data system to build on the successful NHS Numbers for Babies initiative, brought to a halt while the National Programme for IT deals with broader issues.
Newborn bloodspot screening currently has a higher profile than at any time in the past 25 years. Renewed interest by managers and policy makers after years of relative indifference following the abolition of the traditional Health Regions has inevitably caused strains but should, given a period of reasonable stability, enable some of the long-term structural problems in the programme to be tackled. The impact of pathology ‘modernization’ is less predictable and potentially more damaging. Screening laboratories are at the centre of a complex communication web (Figure 1) and have overcome many of the organizational problems affecting pre- and post-analytical phases through direct personal contact. Looking back over the past 35 years it is hard to imagine that there will ever become a time when this role is no longer required. Bridging the disciplines of clinical chemistry and genetics and with a degree of involvement in the ‘patient journey’ that is missing in the more automated laboratory, this is both an intellectually challenging and emotionally satisfying field.
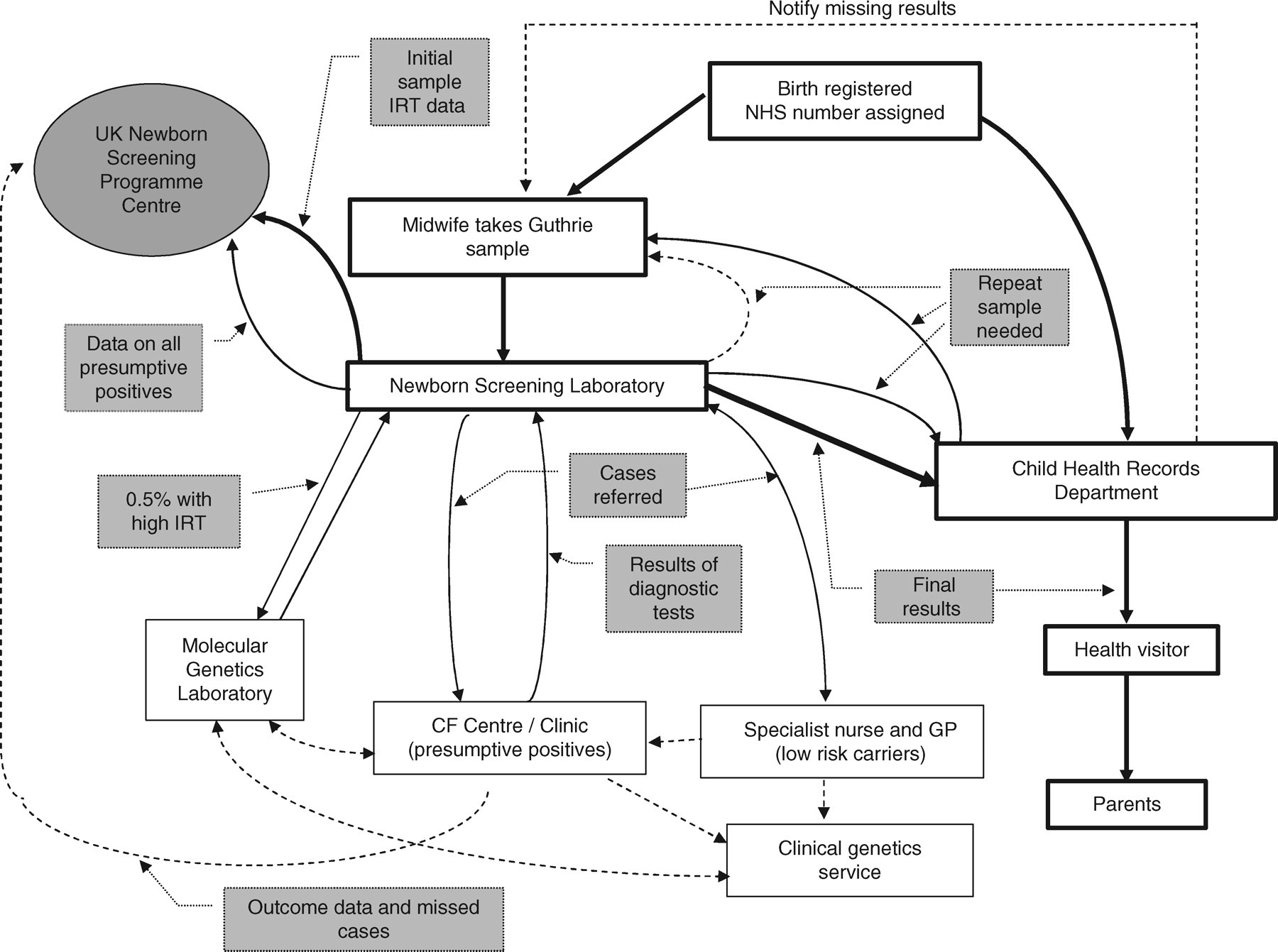
Main information flows in screening for cystic fibrosis