
Abstract
Abstract
Lipoprotein lipase (LPL) is the key enzyme in the catabolism of triglyceride-rich lipoproteins in the circulation. Familial LPL deficiency is characterized by hypertriglyceridaemia and absence of LPL activity. We report a case of LPL deficiency in a 43-year-old woman, who initially presented in childhood with chylomicronaemia syndrome. At that time, her plasma triglyceride concentration was ∼30 mmol/L and post-heparin lipolytic activity was very low. In addition to having the known missense mutation LPL G188E, the patient was also found to have a novel nonsense mutation in exon 8, namely LPL W394X. The novel substitution in exon 8 (c.1262G > A) predicts a truncated protein product of 393 amino acids that lacks the carboxyl-terminal 12% of the mature LPL. Trp394 is part of a cluster of exposed tryptophan residues in the carboxyl-terminal domain of LPL important for binding lipid substrate. Of 11 members from her three-generation family, three were heterozygotes for G188E (mean plasma triglyceride, 3.5 ± 2.0 mmol/L), whereas six were heterozygotes for W394X (triglyceride, 4.3 ± 1.8 mmol/L). In summary, we describe a case of familial LPL deficiency caused by compound heterozygosity for known (G188E) and novel (W394X) LPL gene mutations.
Introduction
Lipoprotein lipase (LPL), a member of the lipase family, is the key enzyme regulating the catabolism of triglyceride-rich lipoproteins in the circulation. Active LPL is a non-covalent dimer of the 448-amino acid LPL protein secreted primarily by adipocytes and muscle cells. 1,2 LPL has an absolute requirement for apolipoprotein (apo) C-II in activating its catalytic centre, which consists of three crucial amino acids, namely Ser132, Asp156 and His241. 1 LPL binds to heparan sulphate proteoglycans on the luminal surface of the endothelium, extending the molecule into plasma where it can sequester chylomicrons and very low-density lipoprotein particles. 1 Lipolysis occurs at the lipid/water interface where substrate access involves a lipid-binding domain in the distal carboxyl-terminal region of LPL. 3 Administration of intravenous heparin displaces LPL into plasma, allowing measurement of LPL activity and mass.
Familial LPL deficiency is a rare autosomal recessive disorder usually diagnosed in childhood, characterized by marked hypertriglyceridaemia and absence of LPL activity. 1 Clinical manifestations can include abdominal pain, episodes of acute pancreatitis, hepatosplenomegaly and eruptive cutaneous xanthomatosis. Familial LPL deficiency is caused by mutations in the LPL gene on chromosome 8p22. About two-thirds of the ∼100 LPL gene mutations described in humans are missense and occur in exons 5 and 6, 3 generally causing a loss of enzyme activity. Treatment primarily involves dietary restriction of fats sufficient to resolve clinical symptoms.
We report a case of familial LPL deficiency found to be compound heterozygous for two LPL gene mutations.
Case report
As an infant the patient experienced multiple episodes of malaise lasting for several days, accompanied by vague abdominal pain, followed by a rash and bruising. At the age of three years, gross hypertriglyceridaemia and chylomicronaemia were noted, and a diagnosis of idiopathic hyperlipidaemia was made. A low-fat diet was instituted, and although it did not stop the episodes, they decreased with age. By the age of 15 years, the episodes had ceased, but her plasma triglycerides were in excess of 30 mmol/L. At 17, further investigations found her to have a very low post-heparin lipolytic activity. 4 In addition, six of nine members of her immediate family had depressed post-heparin lipolytic activity, suggestive of an inherited condition.
The patient was recently reviewed at a Lipid Disorders Clinic. She was well and described no episodes of abdominal pain consistent with acute pancreatitis or cutaneous manifestations of chylomicronaemia on a low-fat diet and was not on lipid-lowering therapy. Owing to the increased risk of acute pancreatitis, her triglyceride levels were closely monitored throughout her two successful pregnancies.
Fasting blood samples were obtained from the proband and her relatives; none were taking lipid-lowering treatment (Figure 1). Informed consent was obtained from all subjects. DNA was extracted from peripheral blood leukocytes using a standard Triton X-100 salting-out procedure. The LPL coding region and exon–intron boundaries of the subject were amplified by polymerase chain reaction (PCR). Primer sequences were based on those described previously. 5 DNA sequencing reactions were performed using BigDye Terminator chemistry and analysed on an ABI 3100 DNA sequencer.
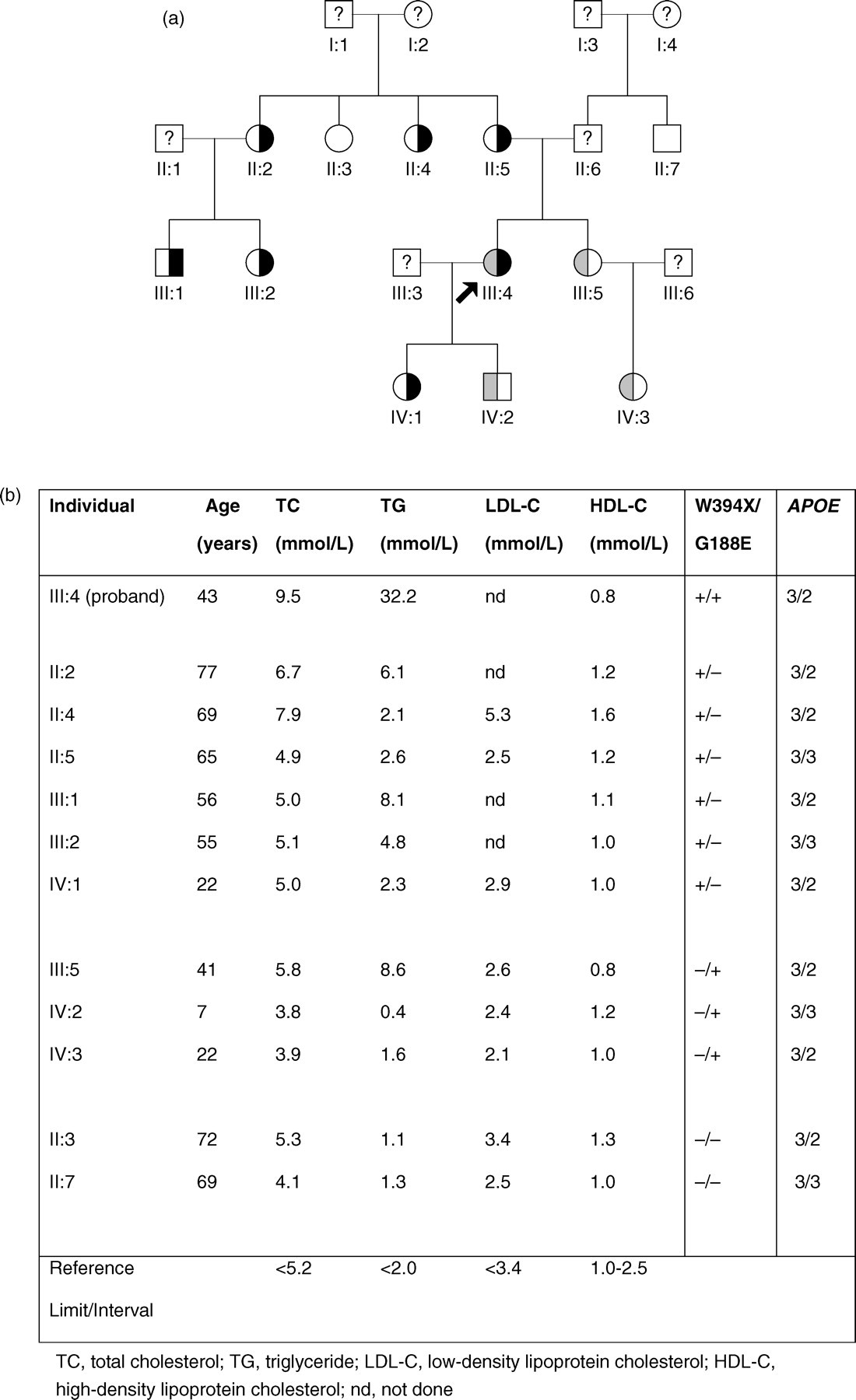
Pedigree and fasting lipid results of the family. (
The patient was found to be a compound heterozygote for two LPL gene mutations. She was heterozygous for the well-studied Gly188Glu mutation in exon 5. She was also heterozygous for a previously undescribed mutation in exon 8, a substitution of A for G at nucleotide position 1262. This nonsense mutation changes tryptophan 394 to a stop codon (W394X). Family studies revealed three members of the three-generation kindred were heterozygous for Gly188Glu (mean plasma triglyceride, 3.5 ± 2.0 mmol/L) and six were heterozygous for Trp394X (mean plasma triglyceride, 4.3 ± 1.8 mmol/L) (Figure 1).
Discussion
We report the case of a patient presenting with LPL deficiency and chylomicronaemia in infancy, who we later found to be a compound heterozygote for a previously characterized LPL mutation, G188E, as well as a novel nonsense mutation, W394X, in exon 8. The novel mutation is predicted to truncate LPL from its expected 448 in the mature protein to 393 amino acids. The loss of the carboxyl-terminal 12% portion of LPL by this mutation occurs at a cluster of hydrophobic tryptophans, namely Trp390, Trp393 and Trp394. Unusually for a hydrophobic cluster, it appears to be located in a hydrophilic region at the LPL protein surface and plays a key role in orienting the enzyme at the lipid/water interface and allowing lipid substrate recognition and access. 6 In vitro studies showed that mutation of Trp393 and Trp394 to Ala completely abolished the ability of the C-terminal portion of LPL to bind to lipoproteins. 7
A wide variation in fasting triglyceride concentrations was observed among obligate LPL mutation carriers in this case study. LPL mutation heterozygotes can be normolipidaemic or exhibit mild hypertriglyceridaemia, 8,9 depending on the mutation present, as well as other genetic and environmental factors such as age, gender, body weight, alcohol intake, smoking and lipid-lowering therapy. 10–12 APOE genotype also influences plasma triglycerides; the ϵ2 allele is associated with higher triglyceride concentrations than the ϵ3 allele. Two-thirds of the heterozygous LPL mutation carriers in this case study have the genotype ϵ3/2, and tended to have higher triglyceride concentrations than their ϵ3/3 relatives.
A patient homozygous for LPL W382X has been described previously; expression studies in COS-1 cells showed that the truncated LPL product (381 amino acids as opposed to 393 in our case) had a marked reduction in expressed mass and had no measurable level of lipolytic activity in cells or medium. 13 These in vitro studies also showed that LPL constructs expressing other C-terminally truncated LPL isoforms ranging in size from 401 to 434 amino acids exhibited no detectable activity. 13 A relatively common LPL variant, S447X, is carried by ∼20% of Caucasians, and truncates the protein by two amino acids. This truncated isoform is unusual in that it is associated with reduced plasma triglycerides, increased HDL-cholesterol, and a decreased risk of heart disease. 14–17 The LPL S447X variant appears to increase LPL expression without changing activity. 18
The plasma lipid profiles in relatives of the proband that are heterozygous carriers of LPL W394X suggests that its impact on LPL activity is equivalent to, if not greater than, that of LPL G188E. Although relatively rare at a carrier frequency of 0.06%, the G188E mutation occurring in exon 5 is the most frequent cause of chylomicronaemia and low HDL-cholesterol in Caucasian populations and has been associated with a five-fold increased relative risk for coronary artery disease. 19 The most common deleterious LPL gene variants, N291S (exon 6, 2–5%) and D9N (exon 2, 1.5%), result in slight decreases in activity without predisposing to chylomicronaemia and have uncertain impact on cardiovascular risk. 3,17,19
In conclusion, we report a case of compound heterozygous LPL deficiency due to the known LPL G188E mutation as well as a novel mutation W394X that truncates the native protein at a hydrophobic tryptophan cluster within the carboxyl domain crucial for lipid substrate binding. The effect of this mutation on the formation, stability and enzyme activity of homo- and hetero-LPL protein dimeric species warrants further investigation.
Footnotes
Acknowledgement
This work was supported by a grant from the Royal Perth Hospital Medical Research Foundation.