
Abstract
Abstract
Background
Familial hypercholesterolaemia (FH) is an autosomal co-dominant disorder which is relatively common, leads to high levels of LDL-cholesterol and if untreated to early coronary heart disease. An audit of current practice at National Health Service Trusts in England was undertaken to determine whether FH patients meet the diagnostic criteria for FH; are being offered appropriate advice and treatment; and to what extent their families are contacted and offered testing for the disorder.
Methods
Medical records of known FH patients (over 18 years of age and diagnosed before 31 December 2003) were accessed to obtain information on diagnosis, treatment and family tracing.
Results
The records of 733 FH patients were examined, 79% met the UK ‘Simon Broome’ register criteria for the diagnosis of definite or possible FH. Analyses showed that patients were usually offered appropriate advice and treatment, with 89% being on a statin. However, the audit indicated a high variability in family tracing between the sites, with significant differences in the frequency of inclusion of a family pedigree in the notes (range 1–71%, mean 35%); the general practitioner (GP) being advised that first-degree relatives should be tested (range 4–52%, mean 27%); and the proportion of relatives contacted and tested (range 6–50%, mean 32%).
Conclusion
FH patients are well cared for in lipid clinics in England, are being given appropriate lifestyle advice and medication, but an increase in recording of LDL-cholesterol levels may lead to improvements in their management. Practice in family tracing appears to vary widely between clinics.
Introduction
Familial hypercholesterolaemia (FH) is an autosomal co-dominant disorder, which is relatively common, having an estimated prevalence in the UK of one in 500 to one in 600. 1 FH leads to high concentrations of LDL-cholesterol from birth and this causes increased risk of premature coronary heart disease (CHD), such that roughly half of men with FH, if untreated, will have developed clinically evident CHD by the age of 55 years. 2 Affected women from the same families typically also develop premature CHD about nine years later than their affected male relatives. 2 Only about 15% of the predicted FH patients in the UK are currently being treated in lipid clinics, 3 with the majority of these being older individuals. 1 Thus most people in the UK with FH are undiagnosed or only diagnosed after their first coronary event, but early detection and treatment with hydroxymethylglutaryl-coenzyme A (HMG CoA) reductase inhibitors (statins) can reduce coronary morbidity and mortality, 4,5 making the identification of affected individuals crucially important.
In the majority of cases FH is caused by a mutation of the LDL receptor (LDLR) gene that impairs its proper function, resulting in very high concentrations of plasma cholesterol. Mutations in two other genes, coding for apolipoprotein B (APOB), the major component of the LDL-cholesterol particle, 6 and for a protease involved in degradation of the receptor (encoded by the PCSK9 gene) also produce the FH phenotype. 7 In the UK, diagnosis of FH is based on criteria developed by the Simon Broome (SB) FH register, 8 which includes LDL-cholesterol concentrations, clinical signs and family history. Currently, in the UK, DNA testing is only undertaken in a few centres, mainly as a result of research projects. 9–11 Modelling has shown that ‘cascade testing’, that is contacting first-degree relatives of FH probands (index cases) and identifying affected relatives by their elevated cholesterol concentrations, is a cost-effective method of finding additional patients. 12 This has been used successfully in Manchester, UK, 13 and extensively in other countries in Europe, most notably in Holland. 14 To examine how applicable this is, the Department of Health has recently funded five pilot sites within England 15 to determine the efficiency of cascade testing within the current social structure and the framework of the National Health Service (NHS).
The first phase of the project, an audit of current practice, was undertaken to determine whether FH patients meet the diagnostic criteria for FH (using the SB criteria); are being offered appropriate advice and treatment; and to what extent their families are contacted and offered testing for the disorder.
Audit methodology
The Department of Health Familial Hypercholesterolaemia (FH) Cascade Testing Audit Project is a pilot project funded by the Department of Health and coordinated by the London and Cambridge Genetics Knowledge Parks in partnership with Heart UK. The coordinating centre was based at University College London (UCL). The two-year project had the equivalent of five full-time nurses based in locations across England (Birmingham, Bournemouth, Guildford, Greater Manchester and Nottingham) involving a total of 11 lipid clinics covering a population of approximately four million. The five locations were chosen from 45 expressions of interest from individual clinics or consortia of clinics. They were selected on the basis that a minimum of 200 FH patients were likely to be available, that the facilities could cope with an increase in workload and that a paediatric service was available. The selection included sites both in and outside large metropolitan areas, and sites with and without a database of FH patients. At each site the FH nurse reviewed all available records to complete the audit questionnaire based on the SB registration form and recorded the data in a Microsoft Access database. Multiple sources of data were used, including the medical notes, letters, local FH databases and the hospital pathology databases. The inclusion criteria for this pre-service development audit was all adult patients (over 18 years of age) diagnosed by clinic staff as having FH prior to 31 December 2003. This cut-off date was selected to ensure that data reflected practices at the clinics prior to their involvement in this project. 16 The coordinating centre was provided with de-identified data.
At one centre, a consortium of four NHS Trusts, it was not feasible to audit all the FH patients within the time available, and 50 patients were selected from each of the four clinics.
Data cleaning
Where information on diagnosis, date of diagnosis, pre-drug treatment lipid concentrations, post-drug treatment lipid levels, gender or medical history was missing, the FH nurse was asked to confirm these data were unavailable. Date fields were checked and corrected. Confirmation was obtained when the numbers of siblings or children recorded were greater than nine, and when the number of contacted relatives was in excess of 19. FH nurses were asked to confirm data for FH patients where noted current lipid lowering therapy did not include a statin. Where patients were not on a statin and no reason was given, the FH nurse was asked to confirm that the reason was not available in the notes. The Friedewald equation 17 was used to confirm that reported LDL-cholesterols were as calculated from noted total cholesterol, triglycerides and HDL. Cholesterol or LDL-cholesterol values >20 mmol/L were checked through several sources.
Determination of diagnosis
The SB diagnostic criteria for FH were used. 8 Patients with a total cholesterol ≥7.5 mmol/L (treated or untreated), or a LDL-cholesterol ≥4.9 mmol/L, were defined as definite FH if tendon xanthomas (TX) were present in either the patient or in a parent, child, grandparent, sibling, uncle or aunt, or as possible FH if there was a family history of myocardial infarction (MI) or raised cholesterol. Patients with known diabetes, renal, thyroid or liver disorders were excluded.
Statistical analysis
All descriptive and statistical analyses were carried out using SAS (9.1.3 Service Pack 3, SAS Institute Inc, Cary, NC, USA). Patients' baseline characteristics and differences observed across the five sites were evaluated. Categorical data were analysed using chi-squared tests; continuous data were analysed using general linear models and Kruskal-Wallis. Concentrations of serum cholesterol, LDL-cholesterol and triglycerides were log-normally distributed; these data were transformed before being analysed using general linear models. P < 0.05 was considered to be statistically significant.
Results
Records of 733 FH patients meeting the inclusion criteria were included. Numbers ranged from 91 to 181 across the five localities. In Table 1, the characteristics of the patients are presented, demonstrating considerable heterogeneity between clinics. Overall, the mean age of patients was 51 years, ranging from 49 to 55 in the different localities, 45% of patients were male (range 38–51%). All 733 records included a medical history containing detailed information on the presence or absence of cardiovascular disease and concomitant treatments. Eighty-nine percent of the patient records (range across clinics 86–96%) had documented evidence of current statin use. Pre-treatment concentrations were recorded for total cholesterol in 705 (96%), LDL-cholesterol in only 301 (41%), with triglyceride in 516 (70%) and HDL concentrations recorded in 424 (58%). Post-treatment LDL-cholesterol was only recorded in 524 patients (75%, range 18–93%). Where both pre- and post-LDL were recorded, the post-treatment level was significantly lower (Figure 1) on average there was a 48% (3.4 mmol/L) reduction in LDL. Sixty-four percent of patients had a greater than 45% reduction in pre-treatment LDL-cholesterol concentrations. Of the 83 patients not prescribed a statin, 59 (71%) had a documented reason for this, including contraindication (15), adverse reaction (13), refusal (10), or use of an alternative lipid-lowering treatment (21), while 24 records gave no information as to why statins were not prescribed.
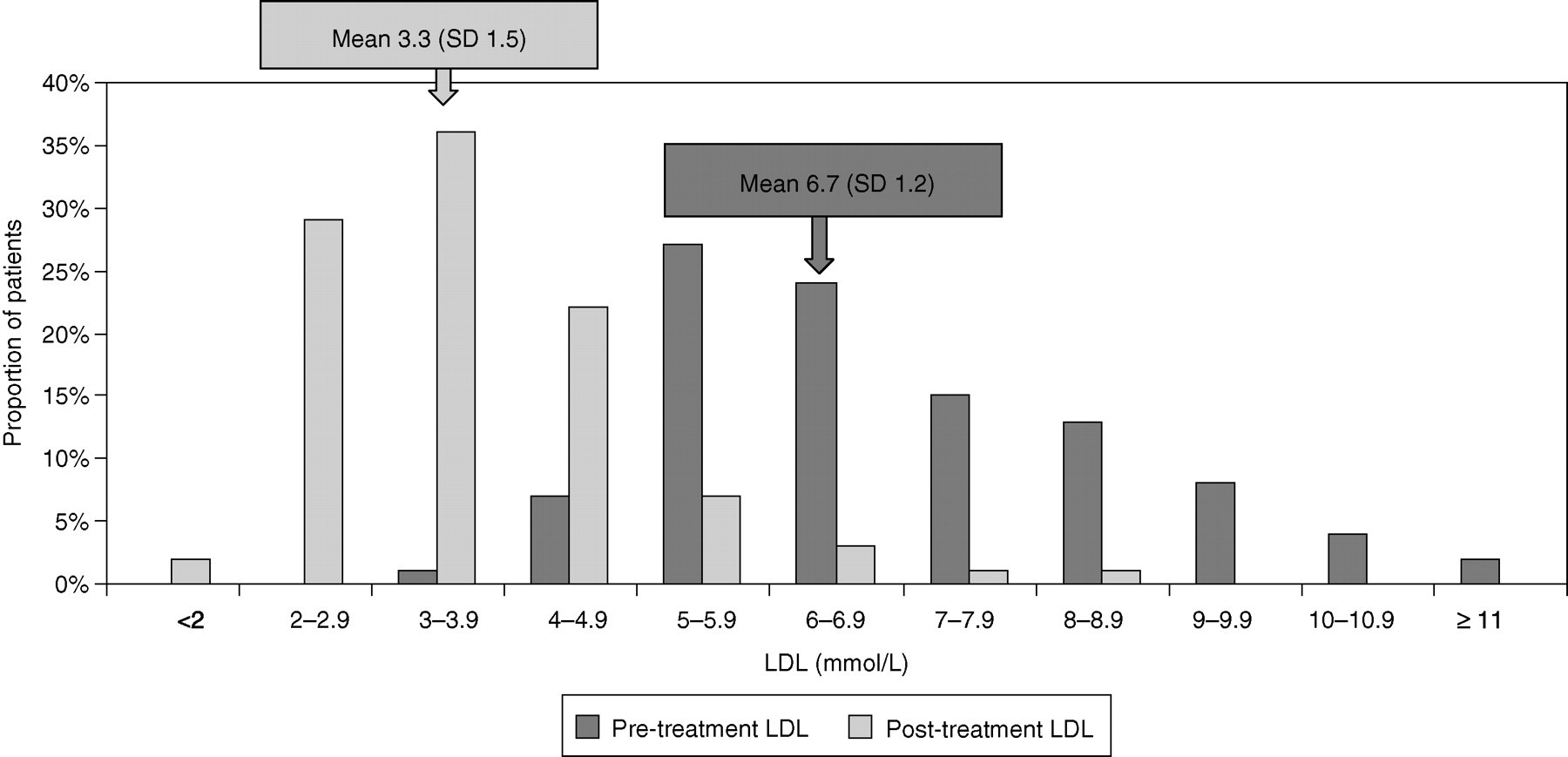
Histogram showing distribution of recorded LDL-cholesterol pre- and post-treatment in familial hypercholesterolaemia patients. P value difference pre-LDL-cholesterol and post-LDL-cholesterol <0.001
Overall characteristics of audit familial hypercholesterolaemia (FH) patients showing the ranges seen in the five locations
*Angina, myocardial infarction, coronary artery bypass grafting
CHD, coronary heart disease
On average, 18% of FH patients had CHD (range 16–26%). The prevalence of TX was also low, at 21% (range 7–37%). However, the majority of records contained documented evidence of a clinical examination for TX (overall mean 75%, range for clinics 28–96%), suggesting that the low value cannot be explained by a failure to look for the physical sign. Other factors explaining this low prevalence were explored. As shown in Figure 2, TX were significantly more prevalent in those with pre-treatment total cholesterol over 11 mmol/L than other groups (P < 0.001). More males than females had TX, and the proportion increased from 11% in those under 35 years to 22% in those over 35 years of age (P = 0.03).
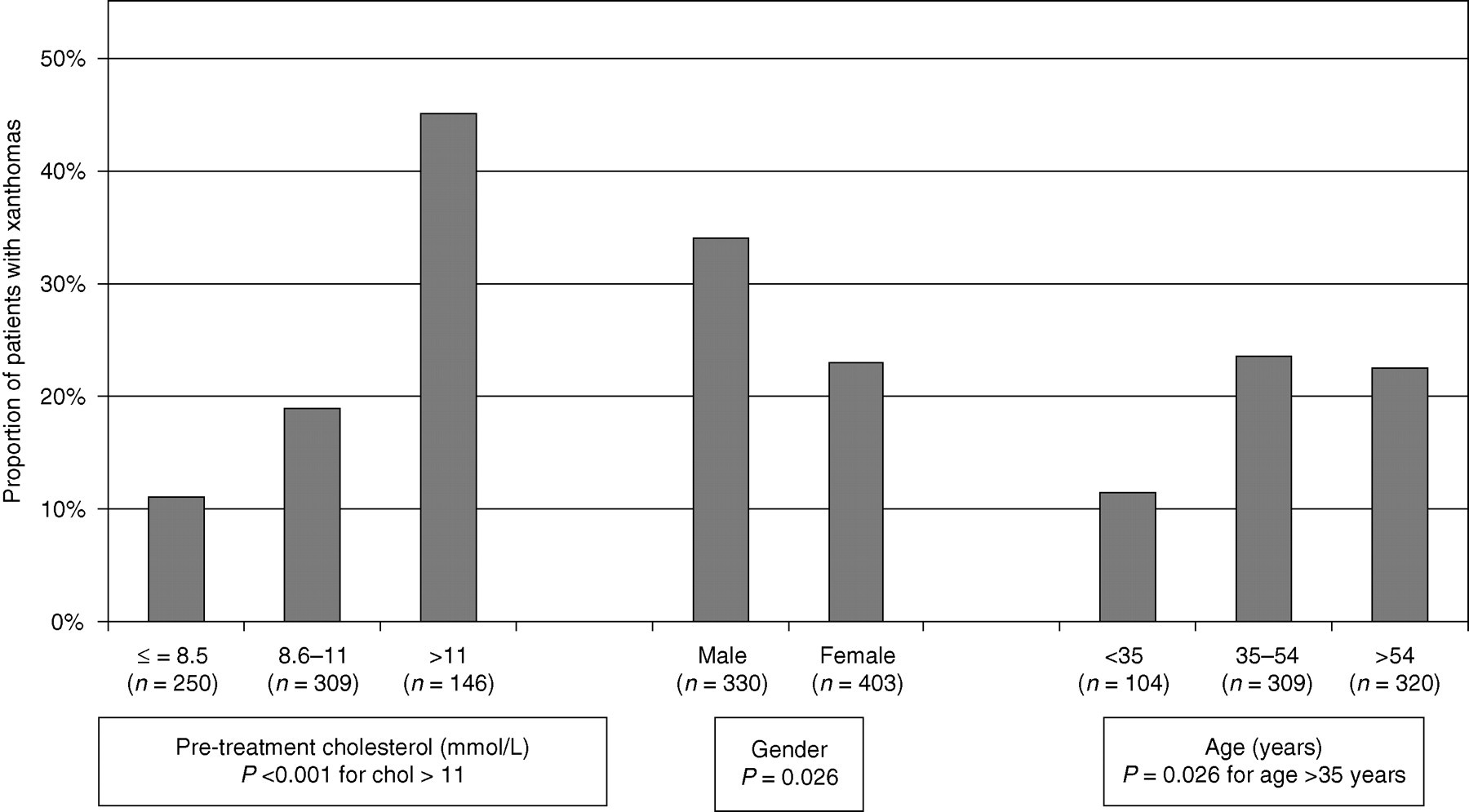
Histogram(s) showing the prevalence of tendon xanthomas in patients by pre-treatment total cholesterol, gender and age
A consequence of the low prevalence of TX was that only 26% (range 8–45%) of the patients had a diagnosis compatible with SB definite FH (a recorded personal or family history of TX). Over half, 53%, had a diagnosis of SB possible FH and for 17% of patients the recorded information was not compatible with either diagnosis, while a further 4% could not be categorized because pre-treatment lipid levels were unobtainable. Four descriptions were found in the patients' records; ‘possible FH’, ‘probable FH’, ‘FH’ or ‘definite FH’, and these were combined into three groups for the analyses (by combining possible and probable FH in the database as possible FH). Data are presented in Figure 3 showing how the diagnosis recorded in the notes compared with the coordinating centre determined, SB diagnosis. In all three groups approximately half the patients were classified as SB possible FH. The proportion of patients meeting SB criteria for definite FH increased from 17% to 46% across the three groups, while those not meeting SB criteria for FH fell from 24% to 6%.
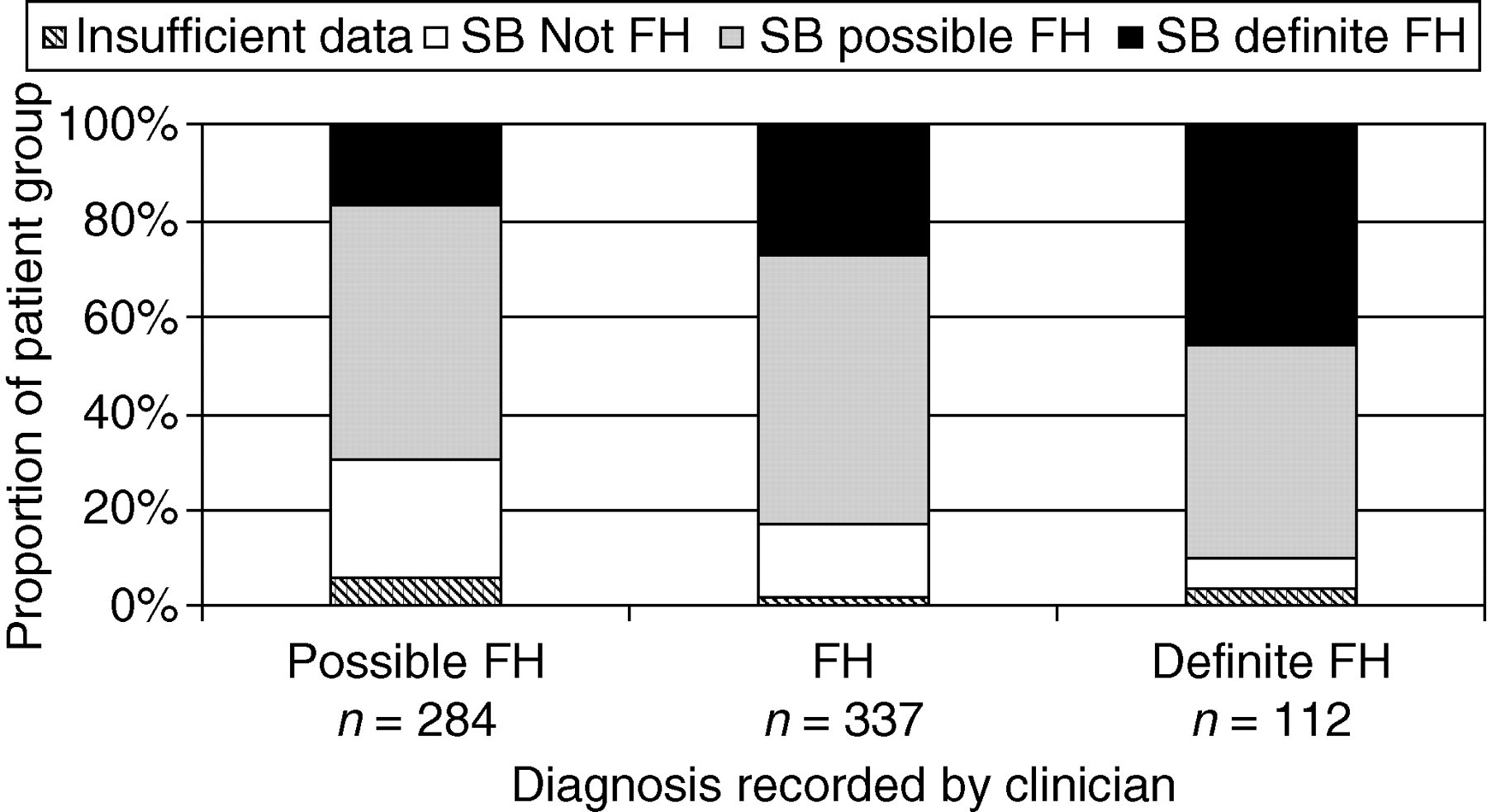
Categorization of familial hypercholesterolaemia (FH) patients: diagnosis as recorded in the notes compared with Simon Broome (SB) diagnosis
Table 2 shows the documented information on the mean pre-treatment plasma lipid concentrations, and post-treatment LDL-cholesterol concentrations in the 705 patients for whom a SB diagnosis could be made . Mean pre-treatment total cholesterol and post-treatment LDL-cholesterol were higher in definite FH. The group of patients that were not FH by SB had significantly higher mean triglycerides than those meeting SB criteria for FH. Mean total and LDL-cholesterol and triglycerides were not significantly different in male and female FH patients, but as expected, HDL-cholesterol was significantly higher (P < 0.001) in female than male patients (data not shown).
Mean (±SD) plasma lipid levels in Simon Broome definite, possible and not familial hypercholesterolaemia (FH) patients at five locations [number]
*Analyses on log-transformed data. Geometric mean and approximate SD shown P denotes difference between the three groups of patients
Since the second phase of the project was to test the first-degree relatives of the FH patients, the audit also searched the notes for evidence of information about the number and status of these relatives; the results are shown in Table 3. Overall, the proportion of notes containing a drawn pedigree was 35% (range 1–71%), and each index case had an average of 4.2 first-degree relatives recorded in the family history (range 3.6–5.3) of which an average of 2.6 (range 1.4–4.0) were recorded to be living relatives. There was evidence that many relatives had already been tested (32%, range 6–50%), with the records showing, on average, 1.4 affected relatives per index case. On average, 27% (range 4–52%) of the letters to the referring general practitioner (GP) contained a recommendation for testing of other family members.
Recorded information about number of tested/not tested and affected/not affected status of first-degree relatives of audit patients
P = significance of difference between clinics. GP = general practitioner
Finally, the notes were examined for evidence of lifestyle advice being given. Overall, 92% (range 74–100%) of patient's notes had evidence that they had been given dietary advice, and 81% (35–100%) of records showed that smokers had been given advice to quit.
Discussion
This audit of the medical records of over 700 FH patients from 11 lipid clinics has demonstrated that, although the proportion on statin therapy is high in all clinics (>85%), there are large differences in many of the clinical, biochemical and family data being recorded. However, since these clinics had actively sought to be involved in the project, and most had a track record in research, it is likely that a similar audit of all UK lipid clinics would find wider differences in the quality of the recorded data. Based on these findings, we propose several ways to improve the recording of data that may have a favourable impact on future patient management.
The clinical diagnosis of FH in the UK is based on the criteria published by the UK SB FH register. 8 This requires the presence of total cholesterol >7.5 mmol/L and particularly of LDL-cholesterol >4.9 mmol/L (in an adult) plus the presence of a personal or family history of TX for a diagnosis of definite FH, or the presence of a family history of early CHD or elevated cholesterol for a diagnosis of possible FH. Using these criteria, 4% of patients could not be categorized, usually because of the absence of recorded pre-treatment lipid concentrations, and a surprisingly high number of patients, 126 (17%), were not classified as FH by SB criteria; usually these had high cholesterol concentrations but no evidence for a familial disorder was recorded. Approximately one-quarter met the criteria for definite FH but this proportion varied considerably between clinics (8–45%). Some differences in patient characteristics are to be expected, since a single clinic serving a large catchment area would be likely to see a broader range of FH patients than one acting as a metropolitan tertiary referral centre, where a higher proportion of more severely affected definite FH patients might be expected. In the absence of a genetic test, the diagnosis of definite FH depends on the identification of TX and even though overall 72% of records show evidence of examination for TX, detection rates are likely to differ between clinicians. However, as reported by others, 18,19 the prevalence of TX in patients increased with male gender, age and pre-treatment cholesterol concentrations, so in part, differences in the TX frequency amongst clinics could be explained by differences in the prevalence of these characteristics.
In general, there was not a good correlation between the recorded ‘definite’ or ‘possible’ diagnosis and the diagnosis according to strict SB criteria (Figure 3). On inspection, this was partly explained by the clinician using data not included in SB criteria, such as evidence of several first-degree relatives being affected with early CHD, or markedly elevated lipid concentrations (e.g. >9.0 mmol/L), in recording a ‘definite FH’ diagnosis. Thus, although historically the identification of TX has been clinically useful in identifying those at highest risk, as patients are identified at a younger age, and with GPs prescribing statins prior to referral, the absence of TX will become less useful in determining the likelihood of whether a particular patient has definite or only possible FH. A diagnosis of ‘definite’ or ‘possible FH’ was only recorded for just over half of the patients (54%), the remainder were diagnosed as ‘FH’. The description ‘probable FH’ was used when consultants were convinced the patient had FH but the patient did not completely fulfil the SB criteria for definite FH, these ‘probable FH’ were categorized with ‘possible FH’. This may explain some of the discrepancies.
In general, the characteristics of the definite and possible FH patients identified in the five centres are similar to those reported elsewhere, although the prevalence of CHD in the clinic patients is lower than expected (average 18%). This compares with the 37% CHD rate in patients included in the Genetic Risk Assessment for FH Trial (GRAFT) study 20 from clinics in the South East of England and 35% in patients from a single clinic in London. 21 The reasons for this are unclear, but seem unlikely to be due to differences in lipid concentrations, since in both of these studies untreated cholesterol concentrations are similar to those seen here (9.8 and 9.9 mmol/L, respectively vs. 10.0 mmol/L here). The lower prevalence of CHD might in part be explained by the younger mean age of the clinic patients (41 vs. 58 vs. 49 years). Another possibility is that some patients with manifest CHD may be being treated elsewhere, for example in cardiology clinics, and may not be being referred to the lipid clinics where the audit was carried out.
Since the diagnosis of FH is based on the presence of elevated LDL-cholesterol concentrations, the accurate recording of pre-treatment lipid concentrations is vital. Thus the absence of a pre-treatment LDL-cholesterol value in more than 50% of subjects is disappointing. The absence of these data may in part be explained by patients being started on statin therapy by their GP prior to referral to a specialist centre. This may be problematic in general practice as LDL-cholesterol concentrations are not measured directly but are calculated using the Friedewald equation requiring a fasting sample to be taken and HDL and triglycerides to be measured. 17 A high proportion of patients' records contain a pre-treatment total cholesterol, HDL and triglycerides, so in the majority of patients a pre-treatment LDL could be calculated. However, UK clinical and laboratory practice has historically been to use total cholesterol rather than LDL-cholesterol concentrations in the diagnosis and monitoring of hypercholesterolaemia, and this may have also contributed to the paucity of recorded LDL concentrations.
Since statin treatment has been shown to be beneficial for FH patients in lipid lowering, 4 slowing progression of coronary artery disease 22 and in reducing CHD mortality, 5 it was expected that the majority of records would document current statin use. This was the case in 89% of records, with no evidence of between-clinic differences. Where statins were not in current use the reason was recorded in the majority of cases. These included adverse reactions, which are rare but can be serious (e.g. rhabdomyolysis 23 ) or rarely, patient refusal. For female FH patients, contraindications include breastfeeding, pregnancy and planning to become pregnant, since statins are associated with risks of congenital defects. 24
Clearly, to monitor effectiveness of medication, details of post-treatment LDL-cholesterol is required, but this was present only in 75% of records (range 18–93%). Where both pre- and post-data were available, overall, treatment had reduced LDL-cholesterol by 3.4 mmol/L with 64% of patients achieving a 45% decrease and just one-third achieving a target of 3 mmol/L. Since the audit, a new lipid-lowering drug, the cholesterol absorption blocker (ezetimibe), has become available, and has been shown to be effective in lipid lowering in FH patients. 25 Thus a new audit might show greater lipid lowering, with more FH patients reaching target. However, a lower target for LDL-cholesterol of 2 mmol/L for individuals at high risk of CHD is now advocated by the Joint British Societies' guidelines (JBS2). 26 At the time of the audit almost all of the FH patients had LDL-cholesterol concentrations over 2 mmol/L, but again, this may have changed since the introduction of ezetimibe, and with clinicians aiming to achieve the new target.
One of the major surprises of the audit data was the extent to which the notes contained evidence of lipid testing in the index cases' relatives. Although only 35% of records contained a drawn pedigree, essentially all records contained evidence that a family history had been taken (as this is required for the diagnosis), and this contained information on over 3000 first-degree relatives, an average of 4.2 per index case with, on average, 1.4 relatives per index case recorded as affected. To some extent this is expected, since evidence of familial transmission is an obligatory part of the diagnosis. Of the approximately 2000 living relatives the notes recorded that overall 32% had been tested. It is relevant that the proportion of the total recorded first-degree relatives who had been tested varied widely between clinics (ranging from 6% to 50%). Whether this reflects differences in clinical practice in contacting relatives or the recording of family information will be determined in the next phase of this project.
Finally, although the notes confirm that most FH patients are being given appropriate dietary and smoking cessation advice, there were differences between clinics. Notably, only in 27% of cases do the records show that the letter to the GP recommended testing of relatives, with again wide differences between clinics (4–52%). Although in cases where the clinician knows that no other relatives are under the care of the referring GP this may be appropriate, generally, making this information more widely included would be a potential benefit. In addition, since FH is inherited as a monogenic disorder, better collection of pedigree and family data would allow identification of at-risk individuals for future testing.
In summary, this audit of notes up to December 2003 shows that although FH patients were being uniformly well-treated with statins and given lifestyle advice, practice varied widely with regards to recording of important biochemical data and pedigree and information on relatives. Local guidelines should specify that the following information be documented for all FH patients: (1) A medical history containing detailed information on the presence or absence of cardiovascular disease and concomitant treatments; (2) A pedigree showing for all first-degree relatives (including those deceased), the date of birth (or age), any CHD and the age at which it occurred, and available lipid data; (3) Post-treatment fasting lipid profiles, including LDL-cholesterol, with pre-treatment concentrations where available; (4) Smoking status, including what advice has been given to smokers; (5) Dietary advice given. The use of the SB registration form is appropriate to ensure these data are consistently recorded.
The notes suggest that each index case has on average at least 2.6 living first-degree relatives and more first- and second-degree relatives may be found if resources such as a nurse were available to take a detailed pedigree. Although a significant proportion of the noted relatives had already been tested, it is likely that further cascade testing would add substantially to the number of FH patients identified in the UK, as suggested recently. 27 Testing this hypothesis is currently underway as the major part of the Department of Health funded project.
Footnotes
ACKNOWLEDGEMENTS
The FH Cascade Audit Project is supported by the Department of Health. SEH acknowledges support from the UK Departments of Health and of Trade and Industry for the IDEAS Genetics Knowledge Park, and from the British Heart Foundation (grants PG2005/014). We would like to thank nurses Ruth Eatough and Fran Lloyd. We also thank the members of the DH project steering group who provided input to the study, in particular Dr R Zimmern, Professor HAW Neil and Dr S Sanderson.