
Abstract
Abstract
Background
Familial ligand-defective apolipoprotein B-100 (FDB) is characterized by elevated plasma concentrations of LDL-cholesterol and apolipoprotein (apo) B, normal triglyceride and HDL-cholesterol levels, the presence of tendon xanthomas, and premature coronary artery disease. FDB cannot be clinically distinguished from heterozygous LDL-receptor-defective familial hypercholesterolaemia (FH) without genetic testing.
Methods
Amplicons in exon 26 and exon 29 of the APOB gene were screened for established genetic variants including mutations and polymorphisms using high-resolution melting analysis. Six novel variants associated with FDB in hypercholesterolaemic Dutch patients (S3476L, S3488G, Y3533C, T3540M, I4350T, G4368D) were also studied.
Results
All positive controls, a total of 10 mutations in exon 26 and four mutations in exon 29, were readily detectable by melting curve analysis. In addition, a patient previously not known to be heterozygous for the H3543Y mutation was identified in a screen of hypercholesterolaemic subjects. The method was validated by comparison of high-resolution melting analysis with DNA sequence data in a ‘blinded’ manner in 35 consecutive patients attending a lipid disorders clinic. These patients were classified as ‘definite FH’ by the Dutch Lipid Clinic Network criteria. Five patients were found to be heterozygous for the R3500Q and one for H3543Y.
Conclusions
We have established a novel, robust method of FDB mutation detection using high-resolution melting analysis in conjunction with DNA sequencing. Compared with existing methods it is not only more cost-effective, but is also capable of detecting new sequence changes and will have importance in cascade screening of affected subjects.
Introduction
Apolipoprotein (apo) B-100 is produced in the liver and is the sole protein component of LDL. 1 The clearance of LDL is dependent on apoB interacting with the LDL-receptor (LDLR). The ability of apoB to interact with the LDLR depends on both sequence and conformation, and is fundamental for the regulation of plasma cholesterol in humans. Defects in the LDLR-binding ability of apoB can cause autosomal dominant hypercholesterolaemia (ADH; OMIM 143890) in the form of familial ligand-defective apoB (FDB; OMIM 144010). 2,3 FDB is characterized by elevated plasma concentrations of LDL-cholesterol and apoB, normal triglyceride and HDL-cholesterol concentrations, the presence of tendon xanthomas, and premature coronary artery disease. The clinical phenotype is a mild form of heterozygous LDLR-defective familial hypercholesterolaemia (FH; OMIM 606945), a similar disorder caused by mutations in the LDLR gene. 4 Other genetic causes of FH include gain-of-function mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) serine protease gene. 5 PCSK9 is secreted and binds to the LDLR extracellular domain, leading to accelerated intracellular degradation of the LDLR. 6 In addition, mutations in an LDLR-adaptor protein that is involved in LDLR internalization cause an autosomal recessive hypercholesterolaemia (ARH), which has a clinically similar, but less severe phenotype than homozygous LDLR-defective FH. 7
The most common FDB mutation involves the substitution of a glutamine for arginine at position 3500 of apoB (R3500Q) and affects about one in 500 individuals of European descent. 8 However, additional mutations in exon 26 affecting surrounding residues (R3480W, R3480P, R3500W, R3500L, R3531C and H3543Y) have been discovered, for which routine screening is not performed. 9–13 These mutations affect the conformation of apoB near the LDLR-binding site, resulting in defective LDL-receptor binding, and an accumulation of atherogenic LDL particles in the plasma. 9 There is evidence for a direct interaction between arginine 3500 and tryptophan 4369 that maintains the correct conformational structure of APOB in LDL, 9 and recently, two mutations in exon 29 associated with FDB have been reported in nearby residues to codon 4369, R4358H and V4367A. 10 Mutations in other exons of apoB, the majority of which are premature stop codons or deletions leading to a truncated apoB protein, result in the opposite phenotype of FH, familial hypobetalipoproteinaemia, characterized by extremely low plasma LDL-cholesterol concentrations. 3,14
Determining FDB mutation status is important, as FDB cannot be clinically distinguished from heterozygous LDLR-defective FH without genetic testing. However, only the most prevalent FDB mutation, R3500Q, is routinely considered, resulting in other causative mutations being undetected and a definite diagnosis of FDB being excluded. This may consequently have implications for treatment of the presenting patient as well as resulting in a missed opportunity for family cascade screening. Traditional FDB genotyping methods such as restriction fragment length polymorphism using mutagenic primers and more recently, 5' nuclease assays, are cheaper and faster than the gold standard, DNA sequencing, but can only detect R3500Q, either alone or in combination with R3500W. 15–17 Other screening methods developed for FDB include heteroduplex analysis, 18 single strand conformation polymorphism, 19 and denaturing gradient gel electrophoresis, 20 but these methods require an electrophoresis step so can be time-consuming and can lack sensitivity. High-resolution melting analysis has recently emerged as a sensitive method for mutation detection. 21 In heterozygotes, the presence of less-stable heteroduplexes changes the shape of the melting curve compared with wild-type homozygotes. These small changes can be reliably detected using a high-resolution melter.
We have developed a closed-tube screening method for FDB mutations and other APOB sequence variations and conclude that high-resolution melting analysis in combination with DNA sequencing is a superior alternative to other methods, with the ability to detect all reported plus novel APOB variants, providing a cost-effective method for the diagnosis or exclusion of FDB.
Materials and methods
DNA preparation
DNA was obtained from consecutive patients referred to the Lipid Disorders Clinic of Royal Perth Hospital, with monogenic hypercholesterolaemia, as well as known FDB patients (determined by DNA sequencing) attending Dutch lipid clinics. 10 All patients were classified as ‘definite FH’ using the diagnostic criteria developed by the Dutch Lipid Clinic Network, as reported by the World Health Organization. 22 DNA was extracted from peripheral blood leucocytes using a Triton X-100 salting-out procedure or from whole blood on an AutopureLS apparatus (Gentra Systems, Minneapolis, MN, USA).
Melting analysis is sensitive to salt concentration, 23 therefore, DNA samples were standardized by sodium acetate/ethanol precipitation followed by resuspension in Tris–EDTA (TE) buffer. In brief, 50 μL of 95% ethanol and 2 μL of 3 mmol/L sodium acetate were added to 20 μL of each DNA sample, vortexed and then subjected to centrifugation (13,500 rpm for 20 min). Seventy percent ethanol was added to the resultant DNA pellet and washed by centrifugation (13,500 rpm for 10 min) and finally resuspended in 50 μL TE buffer.
Polymerase chain reaction amplification
Two sets of primers were designed to amplify all previously reported mutations associated with FDB in the APOB gene. These primers hybridized to regions that do not contain known polymorphisms. These primer pairs are 5′-AGGAGCAGTTGACCACAAGC-3′ (forward) and 5′-TTTGCCATGGAGAGAGTTCCA-3′ (reverse) for exon 26 (365 bp), and 5′-GAAGCTTCTCAAGAGTTACAG-3′ (forward) and 5′-GTCCTTAAGAGCAACTAACAG-3′ (reverse) for exon 29 (156 bp). Each polymerase chain reaction (PCR) mixture contained 1 × EvaGreen dye (Biotium Inc., CA, USA), 2 mmol/L MgCl2, 200 μmol/L each deoxynucleotide triphosphates, 0.125 μmol/L each primer, ∼10 ng of DNA, 250 μg/mL bovine serum albumin (BSA) (Promega), 1 U Perpetual Taq DNA polymerase (Vivantis), 1 × PCR buffer A (Vivantis) and H2O to 10 μL, spun down in a Roche LightCycler capillary. Reactions for exon 26 were overlaid with ∼2 μL of mineral oil to improve accuracy of melting curves.
Rapid amplification was carried out in a Roche LightCycler by initially denaturing at 95°C for 30 s, followed by an optimum 50 cycles of 95°C for 3 s, 60°C for 10 s and 72°C for 15 s. A final extension step of 2 min was performed at 72°C and products were allowed to cool to room temperature.
Melting analysis and DNA sequencing
Before melting analysis, all exon 29 products were centrifuged at 2000 rpm for 5 s, to compensate for any potential effect of increased ionic strength due to evaporation, though omission of this step was found to have an insignificant effect on the curves. This step was omitted for exon 26 products where a mineral oil overlay was used. DNA melting was carried out between 75°C and 95°C for exon 26 and between 70°C and 90°C for exon 29 using the HR-1™ high-resolution melter (Idaho Technology Inc., Salt Lake City, UT, USA) as per manufacturer's instructions. The analysis of the melting curves was performed using the HR-1™ analysis software as provided by the manufacturer. PCR products exhibiting an aberrant melting curve were sequenced using BigDye Terminator V3.1 chemistry (Applied Biosystems, Foster City, CA, USA)-in order to identify and confirm the sequence change. Chromatograms were viewed with Chromas Lite, version 2.01 (Technelysium Pty Ltd, Tewantin, Australia) and analysed using BioEdit Sequence Alignment Editor. 24
Method optimization
It was found that the DNA concentration was not critical to the success of this method, with reliable results being obtained between DNA concentrations of 5 and 50 ng/μL. Whole genome amplification of depleted DNA samples was performed using the Genomiphi™ DNA amplification kit (GE Healthcare, Piscataway, NJ, USA). Material was then diluted 1 in 5 in TE buffer, of which 1 μL was used in each 10 μL PCR reaction. Melting curves generated from PCR products of whole genome amplified material were comparable with the results generated from normal DNA samples. Buccal cell DNA collected on Whatman FTA cards could also be whole genome amplified to generate material of suitable concentration and quality for PCR and melting analysis.
The fluorescent dye EvaGreen was found to give equivalent results to LCGreen Plus (results not shown), the dye typically used in high-resolution melting curve analysis as recommended by the HR-1™ manufacturer (Idaho Technology Inc). However, EvaGreen was chosen in consideration of test economy, as it is a fraction of the cost of LCGreen Plus and all results shown here have been generated using EvaGreen.
The addition of 250 μg/mL BSA to reactions in LightCycler capillaries is recommended by the manufacturers (Roche, Basel, Switzerland), but was found to be unnecessary with comparable results being produced in the absence of BSA.
Results
The current assay was designed to amplify previously reported APOB mutations associated with FDB (Figure 1). Amplicons in exon 26 and exon 29 of the APOB gene were screened for previously established heterozygous mutations (R3500Q, R3500W, R3500L, R3480P, H3543Y, R3531C, R4358H, V4367A) using high-resolution melting. In addition, novel variants associated with FDB and discovered by DNA sequencing in Dutch patients were analysed (Table 1). Melting curves displayed a single melting domain, typically between 83°C and 84°C for exon 26 and 79°C and 80°C for exon 29 in accordance with different product sizes. The melting curves were shifted to the left in the event of a heterozygous sequence variation from the wild-type. Data were plotted as normalized melting curves, difference and derivative plots. Difference plots of normalized data show the difference in fluorescence between each sample and a nominated wild-type control. Derivative plots display the rate of fluorescence change; the peak indicates the melting temperature of a sample. All positive controls, a total of 10 variants in exon 26 and four variants in exon 29, were readily detectable by melting curve analysis and most easily distinguished using the difference plot (Figure 2). To further assess melting analysis, patients carrying two known mutations likely to be encountered more regularly, R3500Q and R3500W, were screened. All heterozygous patients were readily distinguished from wild-type subjects (Figure 3).
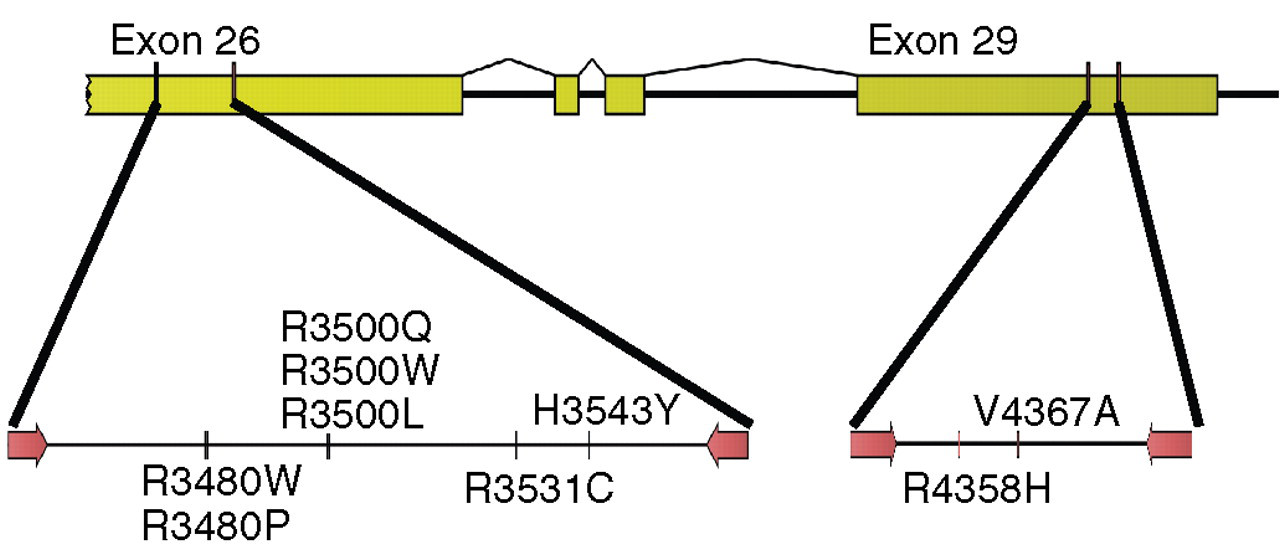
Polymerase chain reaction design to detect familial ligand-defective apolipoprotein B-100 (FDB) mutations by high-resolution melting analysis. Primers (indicated by arrows) were designed to amplify two target regions in exons 26 (365 bp) and 29 (156 bp) of the APOB gene. The positions of all previously reported mutations associated with FDB are shown
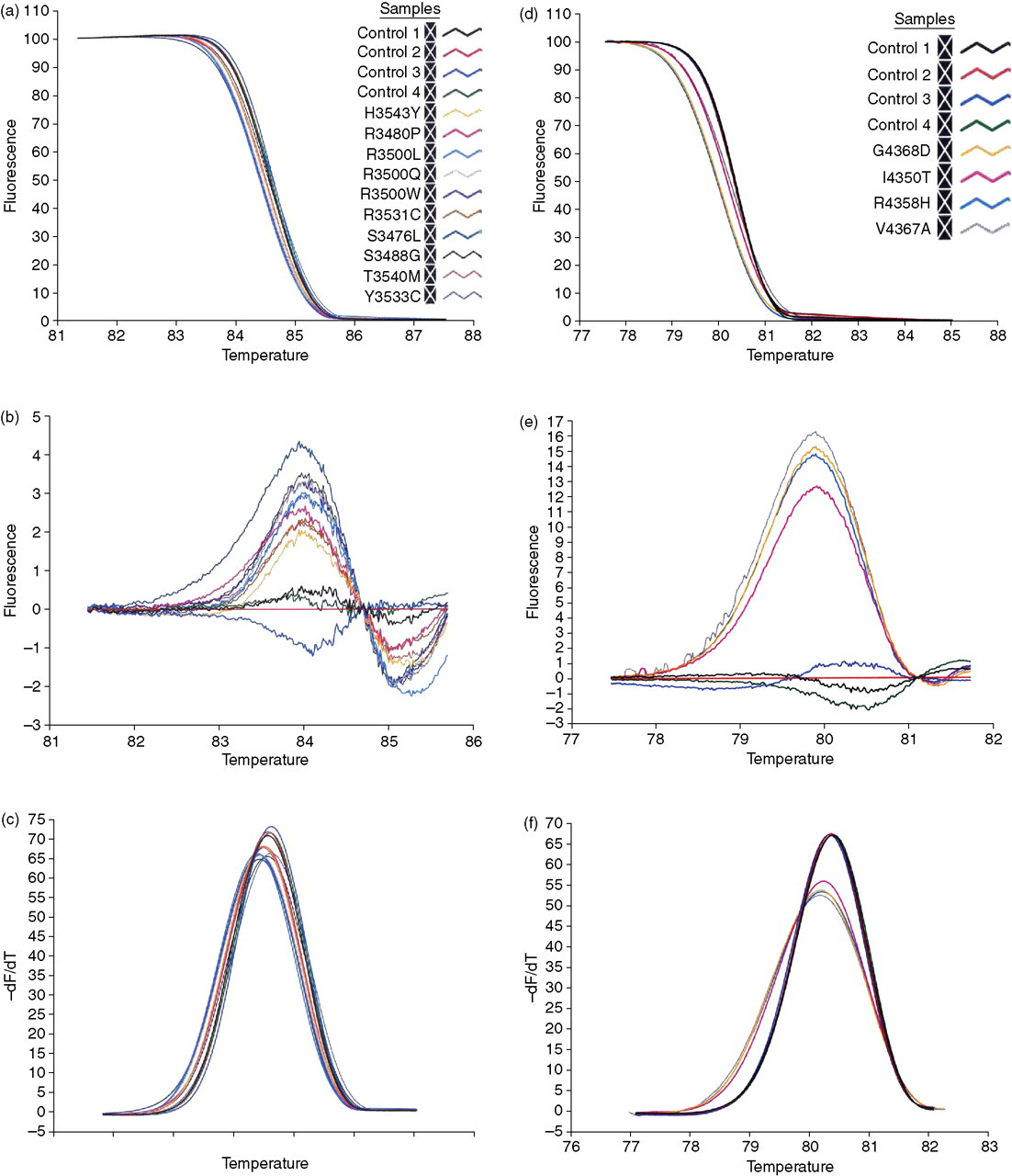
Detection of familial ligand-defective apolipoprotein B-100 (FDB) heterozygotes using high-resolution melting analysis. Previously reported FDB mutations and additional novel mutations (see Table 1) in exon 26 (a–c) and exon 29 (d–f) of APOB were all identifiable using melting curve analysis. The HR-1™ melt analysis software displays results as normalized, difference, or derivative plots. Samples containing heteroduplexes melt at a lower temperature than wild-type and have a normalized melting curve that is left-shifted in comparison with wild-type samples (a and d). The difference plot shows the difference in fluorescence relative to a control sample, and enabled discrimination between heterozygotes and wild-type samples (b and e). The derivative plot of fluorescence with time also distinguished heterozygous from wild-type samples (c and f)
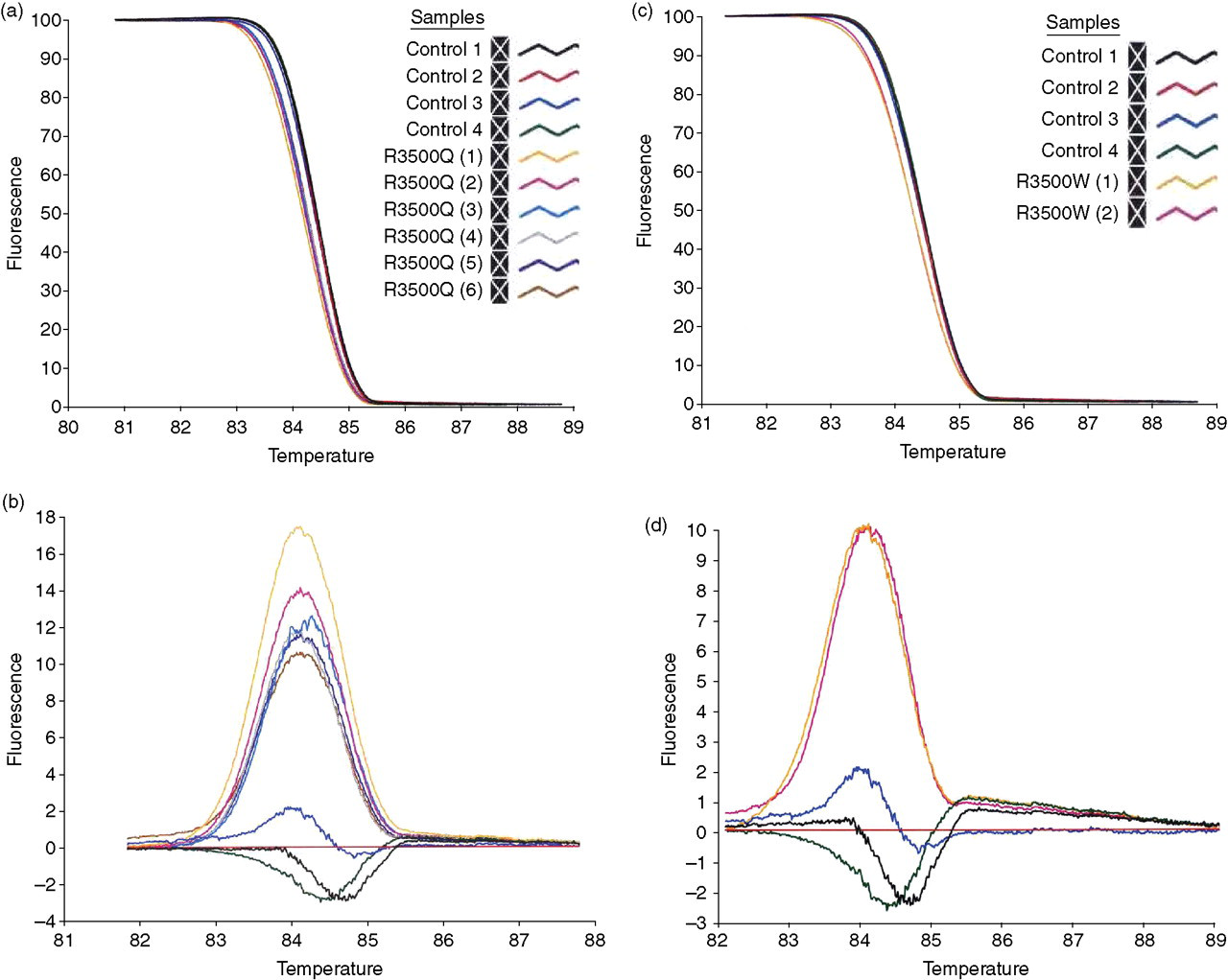
HR-1TM melting analysis of common familial ligand-defective apolipoprotein B-100 (FDB) mutations (R3500Q and R3500W). Melting curves for R3500Q and R3500W heterozygous samples were left-shifted and clearly discernible from wild-type samples following normalization of the raw data (a and c). Difference plots also allowed clear distinction between heterozygous mutant and wild-type samples (b and d)
Novel variations associated with familial ligand-defective apolipoprotein B-100 (FDB) in the Dutch familial hypercholesterolaemia
Reference sequence: GenBank accession no. NM_000384.2. cDNA numbering begins at +1 at the A of the ATG start codon
In a preliminary screen of hypercholesterolaemic patients with suspected FH attending the Royal Perth Hospital Lipid Disorders Clinic, one subject was identified who tested positive for FDB in exon 26 (Figure 4). DNA sequencing revealed that this patient was heterozygous for H3543Y.
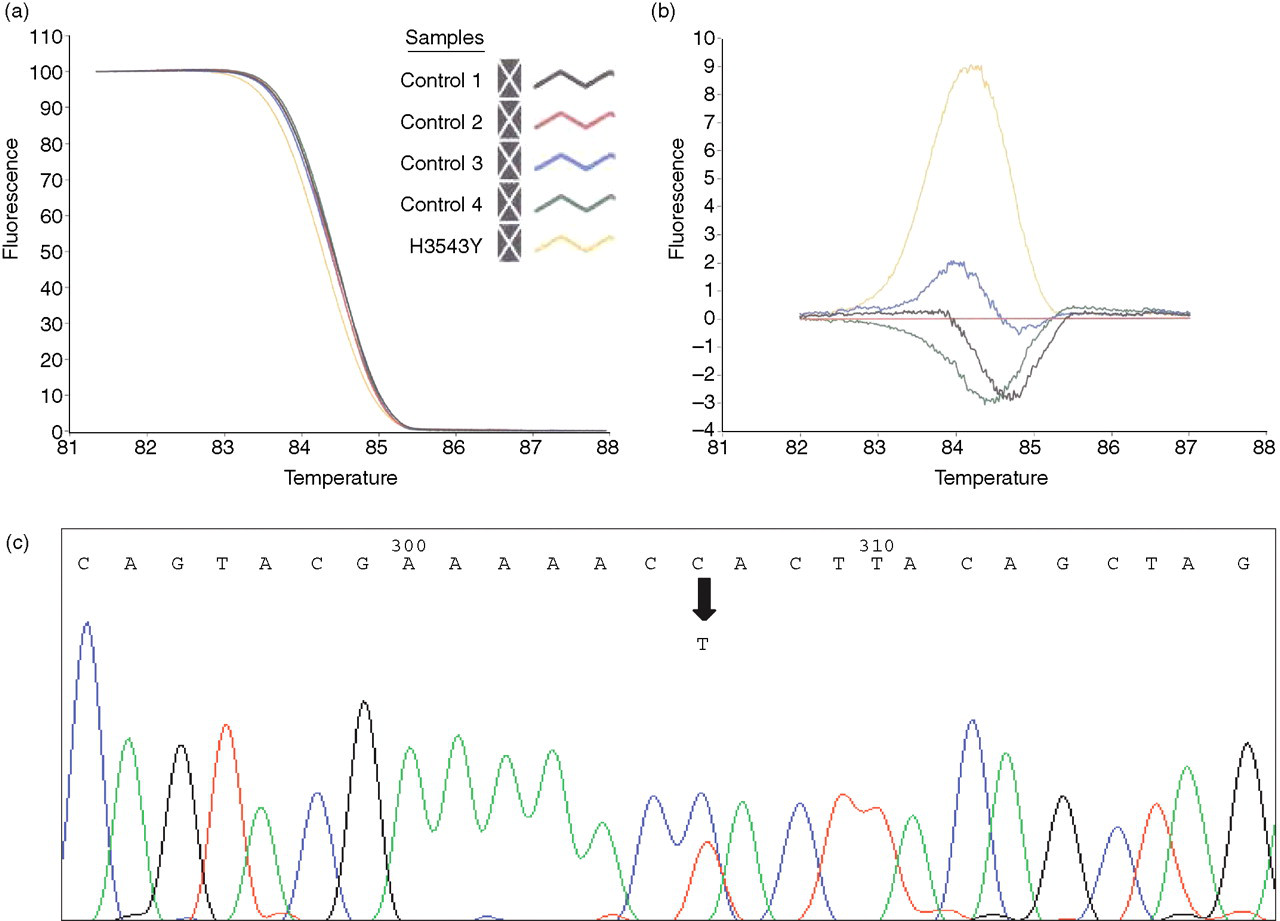
Detection of H3543Y by screening patients using melting analysis. Melting analysis of DNA samples collected from hypercholesterolaemic patients attending the Lipid Disorders Clinic disclosed an aberrant melting curve for one patient. The normalized plot (a) clearly shows a left-shifted curve relative to four wild-type control samples with significant difference in the fluorescence curve (b). Subsequent DNA sequencing revealed a C-to-T change (indicated by the arrow) in exon 26 of APOB, corresponding to H3543Y (c)
To further evaluate this method, a subset of 35 patients attending the Lipid Disorders Clinic were selected for screening based on a definite diagnosis of FH by Dutch Lipid Clinic Network Criteria, 22 with confirmed mutations in either the LDLR or APOB genes. All patient's samples were analysed in a ‘blinded’ fashion, with the mutation status of all patients being revealed by an investigator independent to the study after analysis. Of these 35 patients, six patients displayed an aberrant melting curve for amplicons in exon 26 of the APOB gene and their heterozygous status was confirmed by DNA sequencing. Of these six patients, five were positive for the R3500Q mutation, while one carried the H3543Y variant. The remaining 29 patients displayed melting curves consistent with homozygosity for the normal allele and these results were also confirmed by DNA sequencing.
Discussion
FDB is a common genetic disorder of lipoprotein metabolism, occurring at a frequency comparable with LDLR-defective FH. Unlike LDLR-defective FH, where mutations span the entire LDLR gene, FDB-causing mutations are only found in two specific regions of the APOB gene, making it possible to screen in two segments using high-resolution melting analysis. Fluorescence of a dsDNA-binding dye is detected, and as temperature is increased, DNA strands separate, leading to decreased fluorescence and generating a ‘melting curve’. Heteroduplexes dissociate at a lower temperature than homoduplexes due to non-Watson-Crick base pairing. Analysis and comparison of melting curves allows differentiation of wild-type and mutant DNA samples. DNA sequencing of samples is necessary to identify and confirm the specific sequence change, as the generated melting curves are not necessarily characteristic for a given variant. In addition, DNA sequencing is required to verify that the variation is non-synonymous and likely to cause FDB. A method has previously been developed for FDB mutation detection using realtime PCR and melting curves but used probes and could therefore only detect the FDB mutations at codon 3500. 25
For all products derived from exons 26 and 29, those displaying a ‘normal’ melting profile were subsequently shown to be ‘wild type’ by sequence analysis. All samples with aberrant curves were subsequently shown to harbour sequence variants, demonstrating complete sensitivity and specificity albeit in a test panel of small size. Therefore, we have validated the method by screening, and being able to detect (1) eight previously reported mutations associated with FDB; (2) six novel APOB variants in Dutch hypercholesterolaemic patients and (3) a new case of FDB associated with H3543Y. H3543Y has previously been reported in a German population. 12 Furthermore, in a ‘blinded’ fashion, we have successfully detected sequence variations in a selected cohort of FH patients. Newly detected APOB variants may be rare polymorphisms that do not interfere with binding of apoB to the LDLR. Examination of the families of these index cases by assessing the co-segregation of the newly detected variant with hypercholesterolaemia should quickly resolve the question as to whether or not the new variant is associated with hypercholesterolaemia and is the cause of FDB.
A limitation of our high-resolution melting analysis method for FDB screening is that it would not be able to detect the extremely rare occurrence of homozygous FDB mutations. However, this could be overcome by spiking unknown DNA samples with known wild-type DNA. Using 15% wild-type DNA in unknown samples can distinguish homozygotes from both heterozygotes and normal samples, or 40% wild-type DNA can be used to cluster homozygous mutants with heterozygotes. 26
The HR-1™ was chosen in our study due to its superior performance in comparison with other high-resolution melting systems, 27 and because of its low set-up cost (comparable with a gradient thermal cycler). However, high-resolution melting analysis can be adapted depending on requirements; in other systems 96- and 384-well plates can be used, which would be more suitable for high-throughput applications. Conventional thermal cyclers can be used if a LightCycler is unavailable, and PCR products transferred to capillaries for melting. Melting analysis is cost-effective and quick; analysis only takes 1–2 min per sample, and the major cost in consumables is LightCycler capillaries. PCR and melting analysis occurs in the same closed tube, which reduces the chance of contamination and handling errors.
In addition to being successful for genomic DNA extracted from blood, melting analysis could be obtained using buccal cell DNA on FTA cards combined with whole genome amplification. This is ideal for screening, as samples from remote locations could be collected and sent at ambient temperature through the normal postal system to the laboratory for analysis.
In conclusion, we have established and validated a novel, robust method of FDB mutation detection using high-resolution melting analysis in conjunction with DNA sequencing. Compared with existing methods it is not only more cost-effective, but is also capable of detecting new functional mutations that will have importance in cascade screening of affected subjects.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by a grant from the Royal Perth Hospital Medical Research Foundation. We would also like to thank Dr E Shyong Tai, Department of Endocrinology, Singapore General Hospital, Republic of Singapore for providing us with a R3500W control sample.