
Abstract
Abstract
Background
Urinary 5-hydroxyindole acetic acid (5-HIAA) is a useful marker for the turnover of tryptophan metabolites in the diagnosis and monitoring of carcinoid tumours and the carcinoid syndrome. We have developed a simple and cost-effective assay for urinary 5-HIAA using liquid chromatography–tandem mass spectrometry (LC–MS/MS) incorporating an online sample clean-up process to replace a liquid chromatography electrochemical (LC-EC) technique.
Methods
Acidified urine was serially diluted in ammonium acetate buffer followed by ammonium acetate buffer enriched with 5-hydroxyindole-3-acetic-2,2-D2 acid internal standard. A 2.1 × 10 mm C18 column was used for primary online clean-up and eluted with 100% methanolic mobile phase onto a second dC18 Atlantis 2.1 × 20 mm column. Analytes were detected by mass spectrometry using transitions 192.1 > 146.3 and 194.1 > 148.0 for 5-HIAA and deuterated analyte, respectively.
Results
Run time was 3 min with 5-HIAA eluting at 1.37 min. The inter and intra-assay imprecision and accuracies of the three levels of inhouse quality control (QC) (30, 300 and 600 μmol/L) were acceptable with coefficient of variations (CVs) and deviation from target values <12% (n = 15). The average recovery of 5-HIAA spiked into urine was 93.7% with no ion suppression observed. The limit of detection was 2.8 and lower limit of quantification 4.0 μmol/L. Passing–Bablok regression of LC-EC with LC–MS/MS results showed good agreement between the methods, the relationship described as LC–MS/MS = 1.01(LC–EC)–1.22. No systematic or proportional biases were observed over the working range of the method. The assay was linear to at least 2000 μmol/L.
Conclusions
We have developed a robust method offering a more than six-fold improvement in linearity compared to the existing LC–EC method.
Introduction
Carcinoid tumours are derived from enterochromaffin cells of the gastrointestinal tract and secrete various biologically active amines, proteins and prostaglandins. The symptoms of carcinoid syndrome including flushing, watery diarrhoea, abdominal cramp and bronchoconstriction may manifest when hepatic detoxification capacity is compromised by metastases to the liver. Serotonin is a major product of midgut carcinoid and is derived from hydroxylation and decarboxylation of tryptophan. In health, <1% of available tryptophan is converted to serotonin, which is rapidly sequestered into platelets, while a fraction is metabolized by hepatic monoamine oxidase to 5-hydroxyindole acetic acid (5-HIAA) for urinary excretion. When carcinoid burden is raised, such as in the presence of an aggressive and metastatic midgut tumour, conversion of tryptophan to serotonin is raised and the platelet store of this potent biogenic amine becomes saturated; thus excretion of urinary 5-HIAA is raised beyond the reference range of 10–42 μmol / 24 h. 1
Chromatographic methods coupled to ultraviolet, 2 fluorometric 3 and electrochemical detection systems 4 have largely superseded traditional colorimetric techniques in the routine analysis of 5-HIAA, 5 however, despite their superior analytical sensitivity and specificity, separation techniques of this kind require lengthy run times to achieve optimal chromatographic resolution and detection conditions.
It was felt that the urine catecholamine and 5-HIAA assays remaining on the liquid chromatography electrochemical (LC-EC) system were not cost-effective in terms of the high labour requirement and costs associated with reagent usage and annual flow cell replacement for a relatively small number of samples (approximately 800 samples per annum). In view of our experience with bench-top tandem mass spectrometry, it was decided that a better quality service could be offered by transferral of urinary 5-HIAA analysis to liquid chromatography–tandem mass spectrometry (LC–MS/MS), should it be possible to validate an assay of equal or superior sensitivity and specificity.
The increased selectivity afforded to tandem mass spectrometry, through analysis of specific parent to daughter ion transitions, removes the need for lengthy separations. The implications of this for chromatography are wide reaching. Separation from signal-suppressing matrix contaminants such as proteins, phospholipids or organic ions can be achieved by using shorter analytical columns at relatively high flow rates, greatly reducing the analytical turn-around time for a variety of biological analytes. Recent literature reports the development of LC–MS/MS assays of steroids, 6 immunosuppresants 7 and catecholamines 8 that offer between 5- and 10-fold reductions in analytical run times compared to high-performance liquid chromatography (HPLC) methods employing UV 9,10 or electrochemical detection. 11 It was with this in mind that we set out to develop a LC–MS/MS assay for urinary 5-HIAA to replace the LC–EC etc method.
Methods
A 1 mmol/L stock of 5-HIAA (Sigma-Aldrich, Dorset, UK) was prepared in 0.1 mol/L HCl, and working calibrators were prepared from this by further dilution in 0.1 mol/L HCl to concentrations of 0, 15.6, 31.3, 62.5, 125, 250 and 500 μmol/L. Controls were prepared by spiking acidified urine to 30, 300 and 600 μmol/L from an independent 5-HIAA stock (blank urine 5-HIAA measured 16 μmol/L by LC-EC). The urine was collected into a 4 L 24-h urine container containing 50 mL of 1 mol/L HCl. Further dilutions of QC were prepared to assess the lower limit of quantification (LLOQ). Prior to introduction of the method into the routine workload of the laboratory, Randox urine internal QC material (levels 30 and 300 μmol/L; Randox Ltd., Co Antrim, N. Ireland, UK) replaced the in-house QC material used during validation. Deuterated internal standard (5-hydroxyindole-3-acetic-2,2-D2; CDN Isotopes, Quebec, Canada) stocks were prepared in 0.1 mol/L HCl to a concentration of 5 μmol/L and stored in aliquots at −20°C. Working solutions of 5 nmol/L internal standard were prepared in 0.1 mol/L HCl as needed and stored at 4°C.
Urine samples, controls or calibrators (40 μL) were added to 800 μL 2 mmol/L ammonium acetate, 0.1% (v/v) formic acid (aqueous mobile phase) in microcentrifuge tubes and vortex mixed. This diluted sample/calibrator (20 μL) was further diluted with 400 μL aqueous mobile phase in deep-well microtitre plates containing 20 μL working internal standard, sealed with thermosealing film, mixed, centrifuged at 880 g for 5 min and transferred to the autosampler for analysis. Internal standard was omitted from the first dilution on the basis of economy; given that analyte was not extracted from the matrix, rather, the matrix was extensively diluted, it was expected that losses of analyte during preparation of samples would be negligible.
Online sample clean-up was achieved by injecting 5 μL of prepared sample onto a 2.1 × 10 mm Waters C18 immunosuppressant TDM column, part number 186002695 (Manchester, UK) using a Waters 2795 LC system connected to a PC running Mass Lynx ver 4.0 software (Waters, Manchester, UK). Sample was loaded onto the column and washed with aqueous mobile phase at a flow rate of 0.6 mL/min for 0.5 min before elution at 0.8 mL/min with a step gradient of 100% methanolic mobile phase (2 mmol/L ammonium acetate, 0.1% (v/v) formic acid in methanol), held for 1.5 min. The columns were re-equilibrated to baseline conditions with 100% aqueous mobile phase (2 mmol/L ammonium acetate, 0.1% (v/v) formic acid) for a further 1.5 min at a flow rate of 0.6 mL/min. Further chromatography was performed on a Waters Atlantis 5 μm 2.1 × 20 mm dC18 column positioned between the divert valve and electrospray source of the Waters Quattro Micro tandem mass spectrometer. The mass spectrometer operated in the electrospray positive ionization mode. 5-HIAA and D2 5-HIAA were detected using ion transitions 192.1 > 146.3 and 194.1 > 148.0, respectively. The detector was tuned to the following settings: capillary voltage 0.9 kV; sample cone energy 14 V; collision cell energy 12 eV; collision gas pressure 5.69 × 10−3 mBar; desolvation gas flow 610 L/hr with a dwell-time of 0.05 s/channel.
Routine performance of the assay was assessed several months after introduction of the validated method into the workload of the laboratory. Internal QC and UK National External Quality Assessment Scheme (NEQAS) data for the LC–MS/MS method were compared to results obtained by the LC-EC assay.
Results
Chromatography and linearity
5-HIAA and deuterated internal standard eluted from the Atlantis column at 1.37 min (Figure 1). A peak eluted between 1.40–1.45 min that was visible as an analyte peak shoulder in urines at concentrations near the LLOQ; however, this did not interfere with quantification, as evidenced by the good correlation with the EC method at low concentrations. The total assay time between injections was 3.50 min to allow the solvent gradient to return to baseline conditions. Quantification was performed by integration of the peak area of the extracted ion chromatograms for 5-HIAA and D2 5-HIAA. The calibration curve was linear to at least 2000 μmol/L, with r 2 = 0.997 (n = 5).
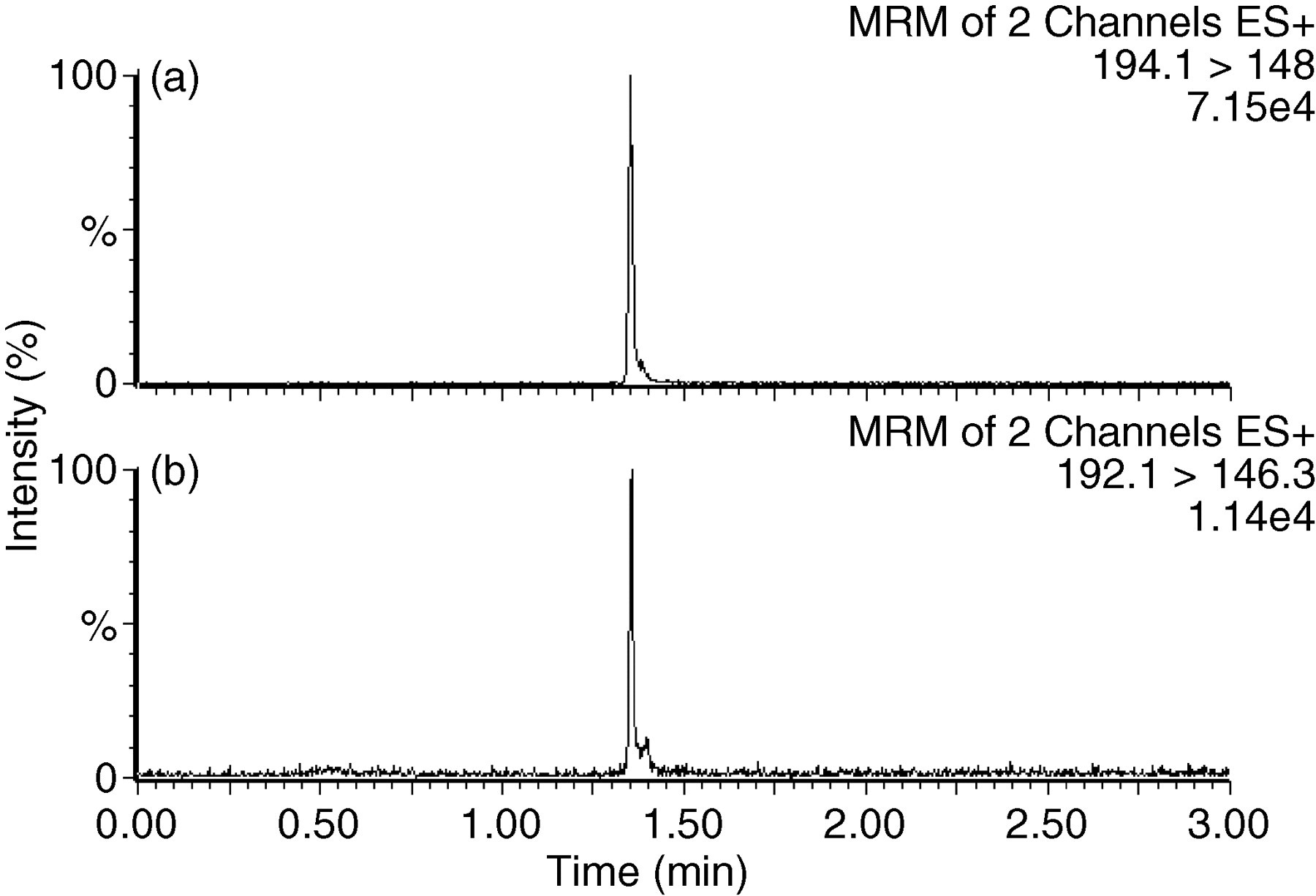
Typical chromatograms of 5-HIAA internal standard (a) and 5-HIAA (b) in a urine sample near the LLOQ (measured value 4.3 μmol/L). Peak area integrated following manual adjustment of baselines for samples at these low concentrations
Ion suppression studies
Ion suppression experiments were carried out with continuous postcolumn infusion of 5-HIAA (100 nmol/L) into the mass spectrometer. Minimal signal interference was noted when extracted samples (n = 6) were injected while 5-HIAA was infused. The main region of ion suppression occurred upon elution of the void volume around 0.3 min with only minor signal suppression noted around the region of 5-HIAA elution. The effect of this was negligible, because the difference between the mean response ratios in urine (n = 6) and aqueous sample (n = 3) enriched to 30 and 300 μmol/L were 2.0 and 6.3%, respectively, indicating ion suppression would not affect quantification of samples against an aqueous standard curve.
Inter and intra-assay imprecision and accuracy
The imprecision of the method was assessed by repeated measurement of the in-house QC material produced specifically for validation described earlier. Samples were analysed in separate batches (n = 15) during a two-week period to calculate between-batch precision, and 12 times within one batch to determine within-batch precision. Interassay coefficient of variations (CVs) and mean percentage deviation from target were <12% for all three levels of QC material. Intra-assay CVs were <6% and deviations from target <15%, acceptable according to FDA guidelines. 12 Since the introduction of this method into the routine workload of the laboratory, the performance of this assay has continued to improve, with Randox urine routine QC CVs of 7.9 and 5.2% with deviations from target values of 10.1 and 1.0% for normal (30 μmol/L) and abnormal (300 μmol/L) concentrations, respectively (n = 17). Two batches of urine 5-HIAA UK NEQAS distributions were analysed by the validated LC–MS/MS method, with favourable returns. The mean positive bias of 17% (distributions 94–96) with the LC-EC method was reduced to a mean positive bias of 8% with the LC–MS/MS (distributions 97 and 98).
Comparison to liquid chromatography electrochemical method
Passing–Bablok regression analysis comparing LC–MS/MS with a LC-EC method showed LC–MS/MS = 1.01, LC-EC = 1.22, r 2 = 0.994, n = 100. Bland Altman analysis showed good agreement of the methods with a bias of 0.23 μmol/L (2 SD limits −37.4–32.2 μmol/L, (Figure 2).
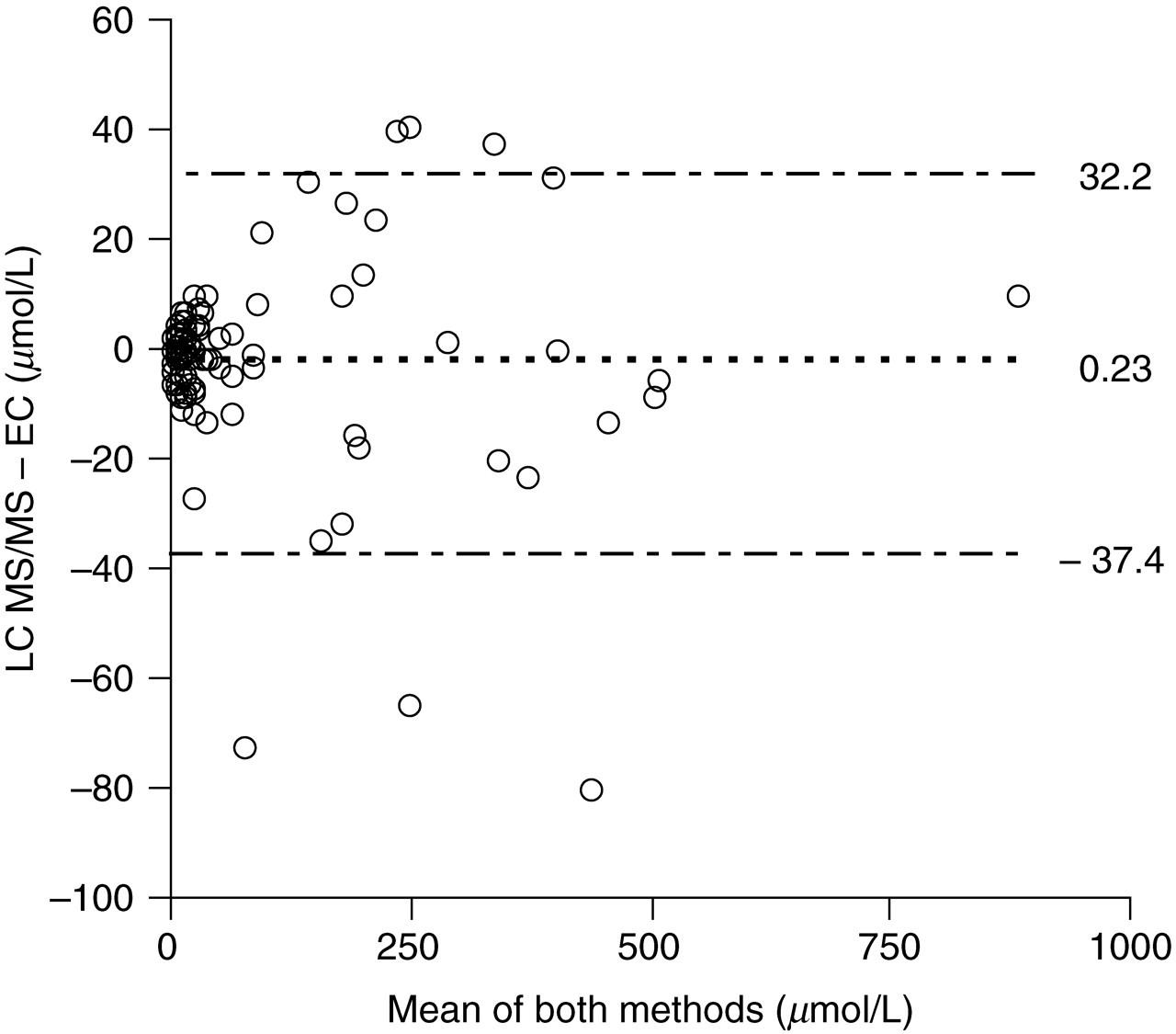
Bland Altman bias plot comparing liquid chromatography–tandem mass spectrometry (LC–MS/MS) to liquid chromatography electrochemical (LC-EC) methods of 5-HIAA measurement in urines of patients with suspected or confirmed carcinoid (n = 100)
Sensitivity and specificity
The limit of detection, calculated as the smallest detectable peak above baseline noise (signal-to-noise ratio >3:1) was 2.8 μmol/L (n = 5). The LLOQ, specified as the concentration with a total CV <15% and mean result with 15% of the theoretical concentration and determined by replicate analysis (n = 10) of control solutions, was 4.0 μmol/L. The mean recovery of 5-HIAA across the range 30–400 μmol/L was 93.7% (range 86.3–100.8, n = 6). Spiking aqueous standards and patient urines (n = 6) with equimolar serotonin or tryptophan did not interfere with measurement of 5-HIAA (data not shown). The acceptability of the results of the validation in terms of accuracy, imprecision, recovery and method comparability confirmed that omission of internal standard from the first dilution stage had not introduced significant error to the assay.
Stability of extracted sample and detector response
The prepared samples were stable for ≥24 h with no significant decrease in concentration (n = 45 external quality assurance samples, paired students t-test, P < 0.01) and detector response over 15 h was stable with CV of response 3.99%.
Discussion
A method for the measurement of urinary 5-HIAA by LC–MS/MS using offline solid phase extraction (SPE) sample preparation has been previously described. 13 Here, we show comparable assay performance with this method, in addition, demonstrating the negligible impact of matrix-induced ion suppression on analyte: internal standard peak height response ratio. Online extraction of 5-HIAA with SPE cartridges on a LC-fluorimetry system has also been reported, 14 demonstrating reproducible online clean-up with up to 40 cycles with a single cartridge. We combine the online extraction concept with sensitive and specific LC–MS/MS detection. We have explored the use of reversed phase C18 analytical HPLC columns for online extraction, further focussing the analyte peak by positioning a C18 column prior to the MS/MS source. No deterioration in column performance has been noted since the urinary 5-HIAA method was introduced five months ago (2000 cumulative injections over 7 months since TDM column replaced). The superior linear range of the LC–MS/MS assay over the LC-EC method reduces the number of repeat analyses of samples measuring over 300 μmol/L and the high sensitivity of the detector permits quantification of small volumes of highly dilute urine samples with accuracy and precision. Extensive dilution of the matrix minimizes on suppression, removing the need for lengthy preparation steps. Owing to the specificity of MS/MS detection, chromatographic resolution of tryptophan metabolites is not necessary, hence, LC–MS/MS analytical run times are greatly reduced compared to LC-EC or fluorimetry.
It has recently been demonstrated that naturally occurring isotopes of cortisol can interfere with MS/MS detection of doubly-deuterated internal standards D2-cortisol. 15 The naturally occurring isotope sharing the ion transition of the deuterated internal standard was detected in the internal standard channel, resulting in a decreased relative response for cortisol and loss of linearity. Data reported here shows no loss of linearity at urine 5-HIAA concentrations as great as 1000 μmol/L, suggesting naturally occurring isotopes of 5-HIAA do not interfere with D2 5-HIAA detection in this instance.
Results from 100 acidified urine samples were compared to results obtained by LC-EC. The results were generally comparable, with the exception of three samples lying beyond the 2 SD limits of the assay (Figure 2). It is possible, these samples had deteriorated in between LC–EC and LC–MS/MS analyses, giving the impression of a substantial negative bias to the LC–MS/MS assay. The equation of the Passing–Bablok correlation did not indicate constant or proportional bias across the working range of the assay. The significance of these three outliers is questionable, given the good agreement between the results from the other 97 samples.
Conclusion
We present a rapid and robust alternative to the LC-EC / fluorometric method for the measurement of urine 5-HIAA which may be of interest to laboratories offering or considering a LC–MS/MS service to local users.