
Abstract
Abstract
A 43-year-old woman presented with a sudden onset of hypokalaemic paralysis requiring intubation and ventilatory support. Subsequent biochemical and clinical assessments established a diagnosis of distal renal tubular acidosis (RTA) in association with underlying Sjögren's syndrome as the aetiology of her profound hypokalaemia. Distal RTA is rare, but Sjögren's syndrome is one of the more common causes in adults and should be considered in the differential diagnosis of patients who present with hypokalaemic muscular paralysis.
Case report
A 43-year-old woman presented to hospital as an emergency with a 24 h history of rapidly progressive limb weakness associated with increasing breathlessness. Her husband had noticed mild weakness of her legs over the preceding week. She had no other systemic symptoms. There was no history of upper respiratory tract, bladder or bowel infection and no recent foreign travel.
On examination, she was tachypnoeic and her speech was feeble. She had difficulty keeping her head upright due to profound neck weakness. Muscle power was reduced in all limbs to grade 2/5. She had a full range of eye movements. There was no sensory loss. The remainder of the clinical examination was unremarkable. She was intubated and ventilated in view of imminent respiratory arrest.
Her past medical history included coeliac disease and hypothyroidism. She smoked 20 cigarettes per day and took alcohol only on an occasional basis. Her medications included thyroxine 150 μg daily, ranitidine 150 mg twice daily, venlafaxine 75 mg daily, tramadol hydrochloride 100 mg twice daily, and lactulose as required.
Although Guillain-Barre syndrome was suspected as the initial diagnosis on clinical findings, biochemical findings were not supportive. These revealed a serum sodium of 139 mmol/L, potassium of 1.2 mmol/L, urea 6.6 mmol/L, creatinine 98 umol/L, bicarbonate 15 mmol/L, chloride 113 mmol/L, magnesium 0.95 mmol/L, calcium 2.08 mmol/L, albumin 31 g/L and phosphate 1.05 mmol/L. Although a metabolic acidosis was present, the calculated anion gap was normal. Urinary pH was 6.73 with an arterial pH of 7.26. Initial urinary electrolyte measurements were potassium 15 mmol/L and sodium 46 mmol/L. Toxicology screen for paracetamol, tricyclic antidepressants, theophylline and salicylate was negative. Full blood picture showed a haemoglobin of 11.7 g/dL, white cell count 14.4 × 109/L and platelets 281 × 109/L. Her electrocardiogram (ECG) had widespread changes typical of profound hypokalaemia (Figure 1a). A central venous catheter was inserted and parenteral potassium replacement instituted with cardiac rhythm monitoring. A total of 1050 mmol of potassium was required to raise the potassium concentration into the reference range. Potassium chloride while initially infused at a rate of 15 mmol/h on admission was subsequently increased, but never exceeded 20 mmol/h (Table 1). She was safely extubated and breathing spontaneously on the second day of ICU admission, but continued to receive corrective potassium supplementation, which allowed her overall strength to gradually improve. The muscle weakness completely resolved with 72 h of intravenous potassium replacement.
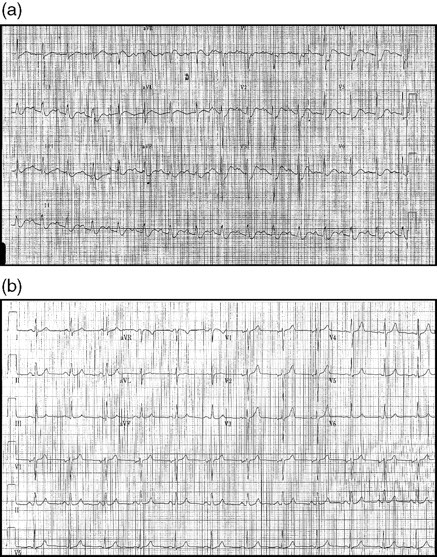
(a) Patient's electrocardiogram (ECG) on admission to intensive care unit (ICU). (b) Patient's ECG after discharge from ICU
Serum electrolytes on admission and over the initial four days during intensive care episode with amount of parenteral supplementary potassium given
The differential diagnosis initially included conditions causing hypokalaemia due to potassium shift from the extracellular space into cells and also those disease states causing overall potassium losses (Table 2). 1 She did not take any sympathomimetic drugs or xanthines known to cause such a cellular shift of potassium, and thyroid function tests performed during her admission were within normal limits. On clinical examination, there were no stigmata of endocrinopathy, and she did not have a family history of familial periodic paralysis. Renal losses of potassium were deemed to be the culprit as no episodes of vomiting or diarrhoea as a potential means of potassium loss from the gastrointestinal tract had occurred. The profound systemic acidosis associated with higher than expected urine pH was in keeping with a renal tubular acidosis (RTA). A bicarbonate loading test was performed while in intensive care by administering 100 mmol of sodium bicarbonate via her central venous line in order to differentiate proximal from distal RTA. Her fractional excretion of bicarbonate was calculated by evaluating the product of the ratio of urinary bicarbonate to serum bicarbonate against the ratio of serum creatinine to urinary creatinine. She had a low fractional excretion of bicarbonate in keeping with a final working diagnosis of distal RTA. After transfer out of intensive care, she remained in hospital until stabilized on a maintenance regimen of oral sodium bicarbonate and potassium supplements. Her ECG changes resolved with correction of her hypokalaemia (Figure 1b).
Differential diagnosis of hypokalaemic paralysis
Adapted from Ahlawat et al. 1
Subsequent outpatient immunological investigations revealed raised anti-Ro and anti-La antibody titres and elevated rheumatoid factor (498 IU/mL). Complement concentrations were within reference range (C3 1.57 g/L and C4 0.16 g/L). She had a hypergammaglobulinaemia (IgG level of 21.90 g/L) with normal IgA and IgM concentrations. On further questioning, she described symptoms compatible with Sjögren's syndrome including a dry mouth, poor dentition and Raynaud's syndrome. A Schirmer's test demonstrated 3 mm of tear flow at 5 min on her right eye and 2 mm on her left eye in keeping with significant keratoconjunctivitis sicca. A final diagnosis of acquired distal RTA secondary to Sjögren's syndrome was made.
Discussion
‘RTA’ comprises a group of disorders characterized by disproportionately alkaline urine in the face of a hyperchloraemic normal anion gap metabolic acidosis. There are three main forms referred to as distal (dRTA, type 1), proximal (pRTA, type 2) and a form secondary to aldosterone deficiency or resistance (type 4 RTA). Type 3 RTA, deemed to represent a combination of the initial two forms, is no longer considered to exist. 2 dRTA is the most common type of RTA and can itself be subdivided into classical, complete dRTA and an incomplete form, not associated with a systemic acidosis, called incomplete or latent dRTA. This patient's dRTA associated with Sjögren's syndrome falls into the ‘classical dRTA’ group. Classical dRTA can be present in up to 40% of patients with Sjögren's syndrome, but is rarely clinically apparent without further specific biochemical investigations. 3–5
In pRTA, urine pH falls in the presence of a very low serum bicarbonate level. In contrast, despite profound systemic acidosis, patients with dRTA have an impaired ability to lower the urine pH below 5.5. 6 A number of dynamic tests are available to classify RTA including the bicarbonate loading, ammonium chloride loading, calcium chloride loading, arginine hydrochloride loading, the sodium sulphate infusion and the furosemide test. 2 The bicarbonate loading test was used in this case. After alkalinization of the urine with bicarbonate loading, the pCO2 of the urine should rise significantly in normal individuals. This is because of enhanced distal H+ secretion, which reacts with the HCO− 3 to form H2CO3. The subsequent liberation of CO2 from this compound permits the subsequent development of a high pCO2 in the tubular lumen. pRTA is characterized by a high fractional excretion of HCO3 − (>15%) as a consequence of reduced ability to reabsorb the large bicarbonate load in the proximal tubule. Patients with dRTA on the other hand, have low values for fractional excretion of HCO3 −. This is a consequence of decreased distal acidification of the filtered bicarbonate subsequently precluding the formation of H2CO3 in the first instance. There are a variety of postulated mechanisms for this characteristic feature including hydrogen ion secretory failure, acid back-leak due to increased permeability to acid, failure to maintain a steep pH gradient between the tubular lumen and blood and an inadequately negative potential difference which therefore fails to facilitate H+ and potassium secretion. 7 It has been shown that H(+)ATPase deficiency is the most commonly recognized defect in patients with Sjögren's syndrome and dRTA. 8,9
Potassium wasting is a characteristic feature of both pRTA and dRTA, but the underlying mechanisms are not fully unravelled. The hypokalaemia has been shown to be at least partly secondary to the primary acidification disorder as it will in fact correct to an extent with alkali therapy. 10 Potassium replacement, the cornerstone of treatment of hypokalaemia in the acute setting, was instituted at less than the recommended 20 mmol/h to avoid rebound hyperkalaemia. 11 It has been estimated that in hypokalaemic states each decrease in total body potassium stores of 100 mmol equates to a reduction in serum potassium of 0.3 mmol/L. 9 This is similar to the findings in this case where the infusion of 1050 mmol of potassium resulted in an increase in serum potassium from 1.2 mmol/L to 4.5 mmol/L (Table 1). This equates to a rise in potassium concentration of 0.31 mmol/L for every 100 mmol of potassium infused.
The potassium wasting seen in dRTA associated with H(+)ATPase pump defect is partly related to the reduction in distal hydrogen secretion. In addition, the collecting tubule intercalated cells have a H(+)–K(+)ATPase pump that secretes hydrogen and reabsorbs potassium. 12 Defects in this pump will reduce H+ secretion and K+ reabsorption favouring a state of potassium wasting. 13
Classical dRTA has multiple aetiologies including primary or idiopathic forms, genetic diseases, autoimmune disorders, dysproteinaemia, hypercalcaemia or hypocalcaemia and exposure to certain drugs or toxins. 2 Autoimmune disorders are most frequently associated with acquired dRTA. 6 The patient whom we describe, having presented in her fifth decade, is more likely to have an acquired tubular dysfunction than a genetic defect. Similar florid presentations of Sjögren's syndrome have been described before with profound hypokalaemia leading to respiratory failure due to muscle weakness. 14–18 Fluorescent immunocytochemistry has demonstrated the absence of H(+)ATPase pumps in the distal intercalated cells in patients with Sjögren's syndrome, supporting evidence for hydrogen ion secretory failure. 8,9 Secretory failure can also prevail, if there is an inadequate potential difference across the intercalated cell. Under normal circumstances, this potential difference is maintained by principal cells, the predominant cell type in the distal tubule. By sodium and water reabsorption and potassium secretion these cells maintain a net balance in favour of a negative intraluminal voltage. This tends to facilitate proton secretion by the intercalated cells. Patients with pure hydrogen ion secretory failure, however, are unable to lower urine pH even when conditions are optimal in terms of an adequate potential difference.
Incomplete dRTA is a milder form of overt classical dRTA. Incomplete dRTA is characterized by an absence of a metabolic acidosis under normal conditions, but an inability to lower the urinary pH after an acid load. A higher urinary NH4 + concentration in the context of any given urinary pH will counteract the failure of other known mechanisms of acid secretion. The excretion of ammonium is indirect involving proximal tubular secretion and subsequent reabsorption in the ascending loop of Henle, diffusion through the interstitium and final trapping in the collecting duct (Figure 2). The additional ammonia is generated as a product of glutamine metabolism allowing for additional acid secretion (in the form of NH4 +) and parallel interstitial bicarbonate absorption (Figure 3). As a result, plasma bicarbonate tends to be in the reference range as opposed to classical dRTA when bicarbonate can fall as low as 10 mmol/L. 6
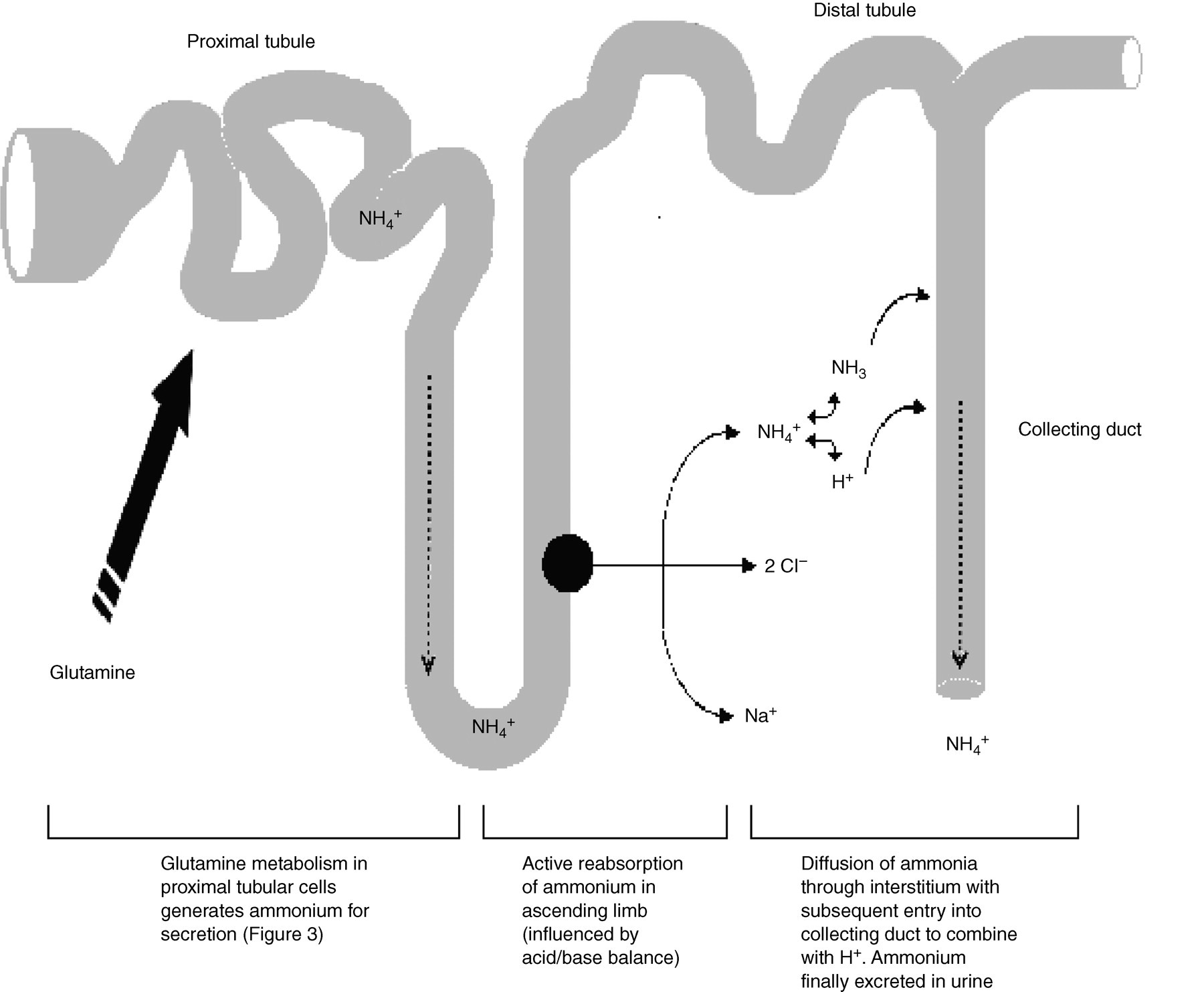
Method of net excretion of ammonium, the key urinary buffer (adapted from Penney et al. 2)
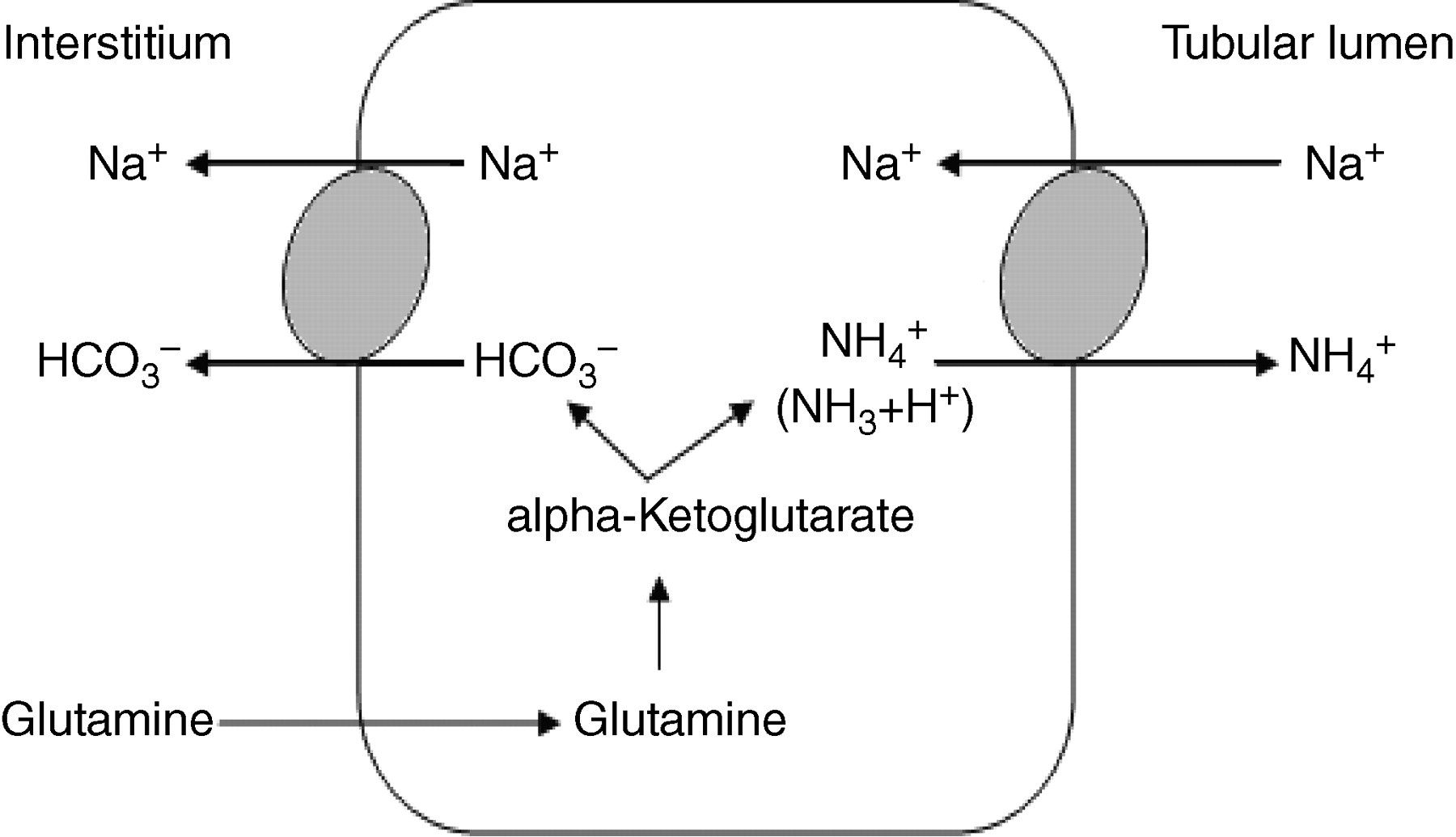
Production of ammonium in proximal tubular cells (adapted from Unwin et al. 6)
Sjögren's syndrome was confirmed in accordance with guidelines established by the American–European Consensus Group. 19 Cases of Sjögren's syndrome presenting in this fashion without the well recognized constellation of symptoms including keratoconjunctivitis sicca, xerostomia, parotid gland enlargement and associated arthralgia are a rare, but previously described phenomenon, which can become apparent even as long as several years after the diagnosis of dRTA (Table 3). 14,15,18,20 These patients with dRTA and associated Sjögren's syndrome tend to be younger (aged 21, 30, 33, 38, 42) 14,16,18,21,22 than those presenting with hypokalaemic paralysis secondary to dRTA without an underlying diagnosis of Sjögren's syndrome (aged 45, 62, 63, 68). 4,15,23,24 This is however not absolute as there is a report of hypokalaemic paralysis caused by dRTA in association with Sjögren's syndrome featuring in as late as the eighth decade. 20 Histopathology from renal biopsy specimens from these patients show evidence of an interstitial lymphocytic infiltrate with plasma cells. Although previous authors have regarded this as contributory to the tubular dysfunction, 20,25 there is not universal agreement with regards to the significance attributed to this. 22 Hypergammaglobulinaemia is often present and has been suggested as a possible pathological means of precipitating distal tubular dysfunction. 3,18 Jordan et al. 26 described a case of transient dRTA in a neonate lasting for 10 weeks, attributed to transplacental transfer of a culprit immunoglobulin from a mother with Sjögren's syndrome. Patients with Sjögren's syndrome, however, can have raised globulin fractions whether or not acidification defects are present. The magnitude of the hypergammaglobulinaemia is independent of any evidence of dRTA suggesting that this frequent finding is not pathogenic in itself. 27 It has also been recently reported that some patients with Sjögren's syndrome have high titres of an autoantibody directed against carbonic anhydrase II. 28 If carbonic anhydrase was inhibited it would result in fewer hydrogen ions being available for secretion.
Learning points from case
Reassuringly, indefinite treatment with oral alkali therapy and oral potassium chloride is very effective in preventing further similar life-threatening episodes of respiratory failure. Follow-up of this patient over a two-year period has shown that she remains clinically and biochemically stable. It was only with the benefit of further careful clinical history, examination and investigation that the underlying primary diagnosis causing her respiratory failure was established.
Footnotes
ACKNOWLEDGEMENT
Apologies to the Authors for the delayed publication, from the Production Team.