
Abstract
Background
Catechol-O-methyltransferase (COMT), an enzyme that metabolizes catecholamines, has recently been implicated in the modulation of pain. Specifically, low COMT activity is associated with heightened pain perception and development of musculoskeletal pain in humans as well as increased experimental pain sensitivity in rodents.
Results
We report that the proinflammatory cytokine tumor necrosis factor α (TNFα) downregulates COMT mRNA and protein in astrocytes. Examination of the distal COMT promoter (P2-COMT) reveals a putative binding site for nuclear factor κB (NF-κB), the pivotal regulator of inflammation and the target of TNFα. Cell culture assays and functional deletion analyses of the cloned P2-COMT promoter demonstrate that TNFα inhibits P2-COMT activity in astrocytes by inducing NF-κB complex recruitment to the specific κB binding site.
Conclusion
Collectively, our findings provide the first evidence for NF-κB-mediated inhibition of COMT expression in the central nervous system, suggesting that COMT contributes to the pathogenesis of inflammatory pain states.
Background
Catechol-O-methyltransferase (COMT) metabolizes catecholamines and thereby acts as a key modulator of dopaminergic and adrenergic/noradrenergic neurotransmission [1, 2]. Converging lines of evidence have revealed an important role of COMT in the etiology and pathogenesis of a wide variety of central nervous system (CNS) disorders [2–4]. Recently, COMT has also been implicated in the regulation of pain perception [5, 6]. Myofacial pain patients exhibit lower COMT activity relative to controls [7], and COMT inhibition increases pain sensitivity in rodents by promoting catecholamine stimulation of β2- and β3-adrenergic receptors [8].
The COMT protein exists in two major forms: a shorter soluble form (S-COMT) and a longer membrane-bound form (MB-COMT). They are encoded from one gene by two mRNA transcripts (1.3 and 1.5 kb in human, 1.6 and 1.9 kb in rats) regulated by the proximal P1 and distal P2 promoters, respectively [9–11]. Only the longer transcript was found in the brain [12] with the predominant protein being MB-COMT. However, S-COMT protein is also expressed in the brain from the longer MB-COMT mRNA isoform via a leaky scanning mechanism [13]. Though their sequences are largely homologous, MB-COMT has approximately a 10-fold greater affinity for dopamine and noradrenaline relative to S-COMT [14]. Seven novel COMT mRNA variants have also been detected in brain, however, they likely to exist at much lower levels than the primary transcript [15]. Although recent reports describe a neuronal expression of COMT [16], it is primarily considered a glial enzyme [17–19].
A significant role for glia in mediating pain has been implicated by studies of patients with persistent pain conditions and animal models of pain [20–22]. Proinflammatory cytokines are produced and released by activated microglia and astrocytes in the CNS as well as by immune cells at the site of injury or inflammation. [23–26]. TNFα is widely considered to be the prototypic proinflammatory cytokine due to its principal role in initiating the cascade of cytokines and growth factors involved in the inflammatory response [20]. Tissue levels of TNFα have been correlated with pain report in a number of painful diseases [27–29]. TNFα activates NF-κB, which is the pivotal regulator of cellular inflammatory responses [30–32]. Specifically, the NF-κB pathway plays one of the major roles in injury or inflammation-evoked activation of astrocytes [23, 33, 34]. Within the nervous system, NF-κB is most frequently composed of two DNA-binding subunits (p65/Rel A and p50) that form a complex with the inhibitory subunit IκB which normally retains NF-κB within the cytoplasm of unstimulated cells [35]. Signal-induced phosphorylation, ubiquitination, and degradation of IκB triggers NF-κB nuclear translocation and DNA binding. Phosphorylation of IκB is mediated by the IκB kinase (IKK) complex, which consists of two catalytic subunits, IKKα and IKKβ, and the regulatory subunit IKKγ [36]. Gene knock-out studies have established an essential role for IKKβ in TNFα-induced activation of NF-κB [37].
A growing number of reports reveal a crucial role of NF-κB in nociception. NF-κB activity is increased in animal models of neuropathic and inflammatory pain [38–42]. A specific IKK inhibitor reverses heightened pain sensitivity to noxious (hyperalgesia) and normally innocuous stimuli (allodynia) [43]. Increased neuropathic and inflammatory pain is suppressed by pretreatment with an NF-κB inhibitor [39, 44]. Interestingly, selective inactivation of NF-κB in glial cells or astrocytes leads to decreased pain and better functional recovery [45–47].
Despite increasing evidence for an important role of NF-κB in pain regulation, very few studies have addressed the mechanisms whereby this pathway exerts its effects on nociception [38,39,41,43,48]. We hypothesized that NF-κB regulates expression of COMT, an enzyme known to contribute to enhanced pain states. Thus, the present study explored the relationship between the NF-κB pathway and COMT expression in order to gain an understanding of the cellular mechanisms underlying inflammatory pain.
Results
TNF inhibits endogenous COMT expression in astrocytes
To elucidate a potential role of TNFα in regulating COMT expression, we treated rat primary astrocytes with TNFα and measured COMT protein levels using Western blot analysis. A significant reduction in COMT protein expression was observed beginning at 8 h (Figure 1A) with a 60% maximal decrease relative to untreated control (P < 0.05; Figure 1B). Using quantitative real-time RT-PCR analysis, we further demonstrated a down-regulation of MB-COMT mRNA at 0.5 h and 30 h following TNFα treatment (P < 0.01; Figure 1C). This oscillatory rather than linear pattern of MB-COMT mRNA expression is in agreement with previously reported characteristics of NF-κB-mediated gene regulation and has been associated with autoregulation of NF-κB activity, as one of the genes activated by NF-κB is that encoding its own inhibitor, IκBα [49]. Finally, a dye exclusion test verified that the TNFα-dependent down-regulation of COMT was not due to cytotoxic effects as more than 95% of cells treated with TNFα remained viable (data not shown).
Cloning and structural analysis of the human distal COMT promoter
To study the signaling mechanisms whereby TNFα regulates COMT expression, we cloned the human distal COMT promoter (P2-COMT) which controls transcription of MB-COMT mRNA (Figure 2A). A 1.5 kb DNA fragment corresponding to the previously published P2-COMT sequences [GenBank:
COMT inhibition by TNF requires NF- B activation
Luciferase reporter gene assays were employed to test the effect of TNFα treatment on P2-COMT activity. A chimeric human P2-COMT/Luc construct was transiently transfected into human H4 astroglioma cells. Consistent with the observed down-regulation of endogenous COMT expression, TNFα treatment decreased P2-COMT activity in time-dependent manner. After a 24-hour incubation with TNFα, P2-COMT activity was reduced to 60% relative to untreated control (P < 0.05 and P < 0.001 at 16 h and 24 h, respectively; Figure 2B). To test if the TNFα-mediated inhibition of P2-COMT activity requires NF-κB pathway activation, H4 astroglial cells stably transfected with IκBα super-repressor (SR), a nondegradable dominant-negative inhibitor of all NF-κB complexes, were transiently transfected with P2-COMT/Luc construct and P2-COMT activity was measured during 24 h of TNFα treatment. H4 IκBα-SR cells were no longer sensitive to TNFα-mediated inhibition of P2-COMT activity (Figure 2B).

As NF-κB is generally recognized as a positive regulator of gene expression, we used a reporter vector with a promoter known to be up-regulated in response to NF-κB activation. A luciferase reporter vector containing κB consensus sites from the MHC class I promoter was transfected into H4 astroglioma cells. After treatment with TNFα, the MHC promoter reporter showed a 14-fold increase in expression, demonstrating that the TNFα-mediated inhibition of COMT expression in astroglioma cells is P2-COMT promoter specific (P < 0.05; Figure 2C).
Finally, to test effect of NF-κB pathway on endogenous COMT expression, we also treated H4 IκBα-SR cells with TNFα. Consistent with reporter assay results, TNFα was unable to significantly repress endogenous COMT protein and mRNA levels in H4 IκBα-SR cells (P > 0.05 for protein level and P > 0.05 for mRNA; Figure 3A, B, and 3C).
TNF inhibits COMT via a canonical NF- B activation mechanism
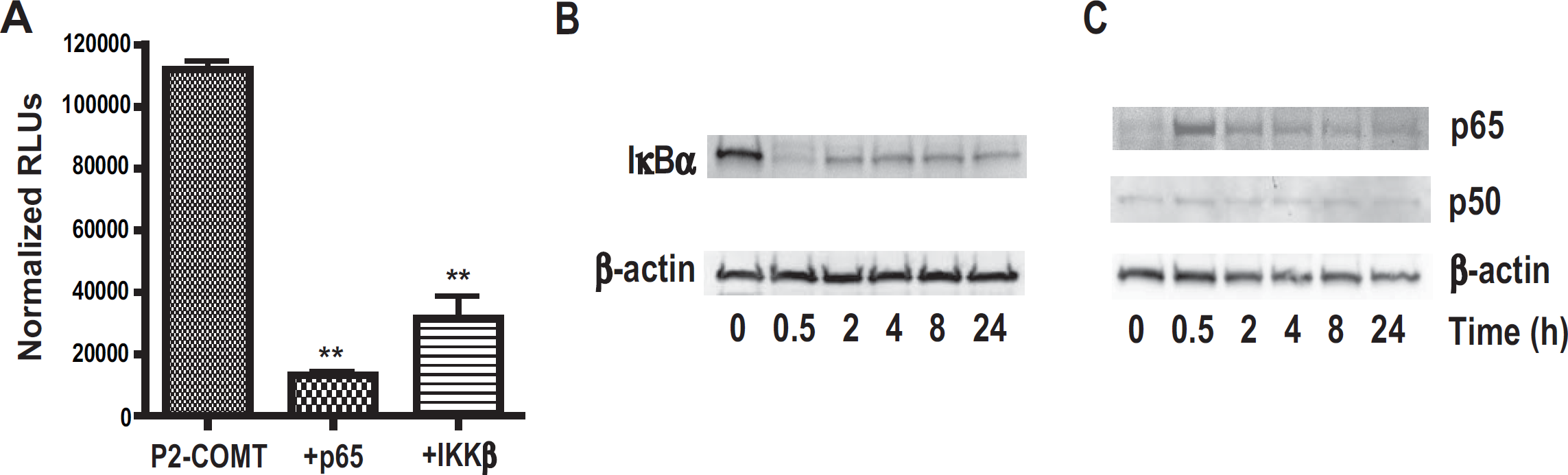
As TNFα may function via NF-κB and JUN N-terminal kinase (JNK) signaling cascades [52], we sought to determine whether a signal that elicits NF-κB activation alone would repress COMT gene expression. Thus, we tested if overexpression of p65 or IKKβ, which are both essential for NF-κB activation, would negatively regulate P2-COMT activity. Our data demonstrate that p65 or IKKβ overexpression dramatically decreases P2-COMT activity (P < 0.01; Figure 4A).
Next, we examined whether TNFα engaged the canonical mechanism of IκBα phosphorylation and degradation to trigger transport of NF-κB to the nucleus in our experimental conditions. H4 cells were treated with TNFα and then cytoplasmic and nuclear extracts analyzed by Western blot. The degradation of IκBα in the cytoplasmic fraction of the cells and simultaneous increase in the nuclear level of p65 was observed at 30 min (Figure 4B and 4C), characteristic of dynamic NF-κB signaling [53]. Together, these results demonstrate that TNFα initiates the canonical IκBα degradation pathway to activate NF-κB in H4 astroglioma cells, and that NF-κB activation inhibits COMT in glial cells.
Identification of the functional NF- B binding site in the P2-COMT promoter
To identify the NF-κB-responsive region in the P2-COMT promoter, we analyzed the activity of serial 5'deletions of the P2-COMT/Luc constructs transiently transfected into H4 or H4 IκBα-SR cells (Fig. 5A). Consequent deletions of 5'fragments led to a graduate increase in overall basal promoter activity in all constructs, suggesting the presence of serial putative negatively regulating elements along the P2-COMT promoter. Del. 2 construct that lack the putative κB consensus binding site also showed a lack of response to TNFα treatment in either H4 or H4 IκBα-SR cells (P > 0.05; Figure 5B and 5C). Conversely, TNFα treatment of H4 but not H4 IκBα-SR cells inhibited activity of construct Del.1 containing the putative κB consensus binding site and the initial P2-COMT promoter construct (P < 0.05 for H4 cells and P > 0.05 for H4 IκBα-SR cells; Figure 5B and 5C). Thus, the region between position −155 and −33 appears to be crucial for TNFα-dependent suppression of P2-COMT activity.

To determine whether TNFα induces NF-κB binding to the consensus DNA site of the identified region, we used ELISA to detect bound NF-κB p65 subunits. This method was employed as an alternative to the electrophoretic mobility shift assay, as it was reported to be more sensitive and quantitative [54]. DNA binding of p65 was dramatically increased in nuclear extracts of H4 cells after 30 min of TNFα treatment (P < 0.001; Figure 5D). This binding was abrogated by incubation of the nuclear extracts with either a competing control wild-type κB consensus oligonucleotide (Figure 5D, WT BC oligo) or with a competing oligonucleotide containing a wild-type putative κB binding sequence 5'-G

Discussion
In the present report, we provide the first demonstration that COMT gene expression is downregulated by TNFα in primary rat astrocytes at both protein and mRNA levels. As the P2-COMT promoter controls expression of MB-COMT, the main COMT transcript in brain, this promoter was cloned from human genomic DNA and transfected in H4 astroglioma cells. The activity of the cloned promoter was substantially suppressed by TNFα in a time-dependent manner.
A number of putative regulatory elements have been described in P1- and P2-COMT promoters, including estrogen response (ER) elements [50] that likely mediate estradiol-dependent downregulation of COMT expression in cell culture [55]. P2-COMT also contains abundant methylation sites associated with cancer development [56], schizophrenia, and bipolar disorder [57]. We identified a novel putative regulatory site – a κB consensus sequence that is a potential target for TNFα-dependent NF-κB activation.
Next, we demonstrated that TNFα-dependent COMT downregulation was indeed mediated by the NF-κB pathway. Transient expression of p65, the essential component of NF-κB complexes, or IKKβ, the major positive regulator of NF-κB activition, significantly decreased P2-COMT reporter expression. In addition, H4 IκBα-SR cells lost the ability to regulate P2-COMT promoter expression in response to TNFα treatment. The TNFα-mediated suppression of endogenous COMT expression was also abrogated in H4 IκBα-SR cells. Moreover, we confirmed that TNFα activated NF-κB in H4 astroglioma cells through the canonical IκB degradation pathway to trigger p65 nuclear translocation and DNA binding.
Our data strongly suggest that the putative κB binding site 5'-GGGGACGCCC-3’ at position −109 of the P2-COMT promoter region is a functional site for NF-κB-mediated regulation of COMT expression as deletion of the P2-COMT region containing this site abrogated TNFα-dependent inhibition of P2-COMT activity in H4 astroglioma cells. Furthermore, competition experiments performed with the wild type or mutant site-specific oligonucleotides showed that TNFα indeed induced recruitment of p65 to this κB consensus binding site of the promoter.
Although NF-κB-mediated activation of transcription is well known, the mechanisms of NF-κB-mediated repression are poorly established. Probably, the best studied example of transcriptional repression by NF-κB complex is described for Dorsal transcription factor, a Drosophila Rel family member that can either activate or repress gene expression through the recruitment of coactivators such as CBP or corepressors such as Groucho [58]. Furthermore, a number of examples have been reported in mammals. NF-κB can repress transcription by competing with steroid receptors for a common promoter cis-DNA element [59]via N-myc recruitment to the glutamate transporter gene promoter [53] and through inhibiting histone H4 acetylation at the cytochrome P-450 1A1 promoter [60]. Thus, further experiments should be conducted to address the specific mechanism underlying NF-κB-dependent inhibition of COMT gene expression. Interestingly, consequent deletions of 5'fragments of the P2-COMT promoter led to a significant increase in overall basal promoter activity. This result would suggest the presence of a number of putative negatively regulating elements along the P2-COMT promoter other than ER- and κB-response elements. Although, to date, no studies have systematically searched for regulators of COMT expression, this finding clearly warrants further research.
Our results demonstrating that COMT expression is downregulated in astrocytes under inflammatory conditions are in line with those of other studies showing a positive correlation between astrocyte activation and exaggerated pain responses [24, 25, 61]. Intrathecal injection of gp120 (human immunodeficiency virus-1 envelope glycoprotein) induces mechanical allodynia via the release of proinflammatory cytokines and NF-κB activation in spinal cord astrocytes, but not in microglial cells or neurons [62]. Selective inactivation of astroglial NF-κB in transgenic mice expressing a dominant negative form of the inhibitor IκBα leads to a dramatic improvement in functional recovery after contusive spinal cord injury (SCI) [46] and decreases formalin-induced pain [47]. Additionally, several recent studies report cell type-specific NF-κB activation by cytokines. For example, in rat brain cultures IL-1 induces NF-κB activation in astrocytes, but not in neurons [63, 64]. Taken together, these studies unequivocally link NF-κB activation in astrocytes to pain states.
Although activation of the NF-κB pathway has been deemed critical for the development of pain [40–42], there are few reports studying NF-κB-dependent pro-nociceptive signaling. Historically, these studies have focused on NF-κB-dependent up-regulation of pro-inflammatory cytokines [25, 65], cyclooxygenase-2 (COX-2) [39, 43], inducible and neuronal nitric oxide synthases (iNOS and nNOS) [38, 41], c-src [48], and c-fos [38]. However, recent studies from our group demonstrated that genetic variants of COMT coding for low enzymatic activity are associated with heightened experimental pain sensitivity and the onset of a myofacial pain condition in humans [5]. Additionally, pharmacologic inhibition of COMT in a rat model of inflammation resulted in elevated pain sensitivity [8]. Together, these data suggest that an NF-κB-mediated decrease in COMT expression is likely to contribute to heightened pain sensitivity under inflammatory conditions. A series of in vivo experiments further addressing this hypothesis are currently being conducted in our laboratory.
Conclusion
Collectively, our results provide the first evidence that COMT expression is downregulated in astrocytes via recruitment of the NF-κB complex to a specific κB-site at the P2-COMT promoter. NF-κB-mediated inhibition of COMT in the CNS may represent a novel mechanism contributing to inflammatory pain. NF-κB is regarded as one of the most important targets for therapeutic intervention against inflammatory conditions [66, 67]; thus, elucidating the cellular mechanisms that underlie NF-κB-mediated inflammatory pain will promote the development of novel therapies including pharmacologic agents that block COMT-dependent pain signaling.

Methods
Cell culture and reagents
Primary astrocytes were isolated and cultured as described earlier [68]. Human H4 astroglioma cells were obtained from ATCC (HTB-148) and cultured in DMEM (Sigma), 10% fetal bovine serum (FBS; HyClone) and 1x penicillin-streptomycin (Invitrogen). H4 cells stably expressing IκBα-SR were a generous gift from Dr. Baldwin (UNC) and generated as described previously [53]. All oligonucleotides were obtained from MWG-Biotech AG. The pCMV-SPORT-M, pCMV-SPORT-M-p65 and pCMV-SPORT-M-IKKβ expression vectors were a generous gift provided by Dr. Romanov (Attagene) and 3x-κB/luc construct was a gift from Dr. Baldwin (UNC).
Quantitative real-time RT-PCR
Total RNA was isolated using the Trizol reagent (Invitrogen), treated with RNase free-DNase I (Promega) and reverse transcribed with random primers by Superscript III (Invitrogen). The cDNA was amplified with SYBR Green PCR master mix (Applied Biosystems) using forward and reverse PCR primers (5'-CCAGAGGAGACCCCAGACC-3’ and 5'-ACAGCTGCCAACAGCAGAG-3', respectively, for human MB-COMT; 5'-GGAAATCGTGCGTGACATC-3’ and 5'-CATGGATGCCAAGGATTC-3', for human β-actin; 5'-CCAGAGGAGACCCCAGACC and 5'-ACAGCTGCCAA CAGCAGAG-3', for rat MB-COMT; and 5'-TGCGGGTCATAAGCTTGC-3’ and 5'-CGATCCGAGGGCCTCACTA-3’ for rat 18S rRNA) in S2 Real Time PCR machine (Eppendorf). PCR reactions were performed in triplicate. Three independent experiments were performed, and the result of a representative experiment is shown. MB-COMT mRNA levels were normalized to β-actin RNA or 18S rRNA as an endogenous control.
Western blot analysis
10–50 μg of protein lysates from whole cells, nuclear and cytoplasmic extracts, normalized for protein content using a BCA Protein Assay Kit (Pierce), were run on precast Novex Tris-Glycine gels (Invitrogen), blotted onto nitrocellose (Whatman), and blocked in TBST with 5% nonfat dry milk. The following antibodies were used: COMT (Chemicon, AB5873), β-actin (I-19) (Santa Cruz, SC-1616), IκBα (C-21) (Santa Cruz, SC-371), p65 (Cell Signaling, #3034), and p50 (Santa Cruz, SC-7178). Chemiluminescence was detected in ImageQuant-ECL Imaging System (GE Healthcare) and images were analyzed using ImageQuant TL software (GE Healthcare). Blots from three independent experiments were densitometrically analysed and the values normalized to the β-actin control, with untreated group set to 100%.
Cloning of human P2-COMT distal promoter
Primers
Construction of serial 5'-end deletions of human P2-COMT/Luc clone
Serial deletions were generated by PCR amplification of corresponding fragments from P2-COMT/Luc clone using forward primers, containing Mlu I restriction site, and reverse primers, containing Bgl II site,
Transient transfection, luciferase and -galactosidase assays
Cells were seeded into 12-well plates (5 × 104cells/well) and transfected with 500 ng of total DNA using FuGene 6 reagent (Roche). Normally, up to 400 ng of P2-COMT luciferase reporter and 30 ng of control plasmid for transfection efficiency (pSV-β-galactosidase vector, Promega) were used for transfection. The amount of DNA was kept constant by addition of pCMV-Sport-M vector with no insert. Cells were treated by TNFα (R&D Systems) and harvested 48 h after transfection. Luciferase activity was determined using Luciferase Assay System (Promega) and normalized for transfection efficiency by measuring the β-galactosidase activity using a β-Galactosidase Enzyme Assay System (Promega). Transfections were performed in triplicate, and a representative experiment is shown.
ELISA for activated NF- B
NF-κB activation was measured using TransAM NF-κB p65 Chemi Kit (Active Motif). Cell lysates were tested for their ability to bind to a plate-immobilized oligonucleotide containing a κB consensus binding site (5'-GGGACTTTCC-3'). Competition experiments were performed with the wild-type (ACCGC
Statistical Analysis
Protein, mRNA, and promoter activity data were analyzed by paired t-test and analysis of variance (ANOVA) with post-hoc tests. P < 0.05 was considered to be statistically significant.
List of abbreviations
COMT: catechol-O-methyltransferase; IκBα: inhibitory factor ΚB; IKK: IκB kinase; NF-κB: nuclear factor κB; TNFα: tumor necrosis factor α.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IET and LBD conceived of the study, and IET, AGN, SW, MC, and LQ performed experiments. IET, AGN, and LBD participated in writing of the manuscript. All authors read and approved the final manuscript.
Footnotes
Acknowledgements
We thank Drs. Maixner and Baldwin for helpful and inspiring discussions, Kathryn Satterfield, Brad Cooke, and Dustin Gibson for technical assistance, Dr. Sitcheran for helpful discussions and a gift of H4 IκBα-SR cell line and B reporter, and Dr. Romanov for p65 and IKKβ expression vectors. This work was supported by the NIH/NIDCR RO1 DE016558 grant to LBD and the NIH/NICHHD K12 HD052191 grant to AGN.