
Abstract
Cardiac rhabdomyomas are the most common fetal cardiac tumor. They can be detected in the second and third trimesters. Rhabdomyomas are most commonly associated with the genetic disorder tuberous sclerosis complex. When associated with tuberous sclerosis complex, cardiac rhabdomyomas usually regress within the first few years of life, without complications. Symptoms depend on the size, number, and location of the rhabdomyomas. A case report of multiple cardiac rhabdomyomas that was found at 35 weeks’ gestation and is discussed.
Fetal cardiac tumors are rare. Of these tumors, the most common are rhabdomyomas; less often, fetal cardiac tumors are diagnosed as fibromas or teratomas. Cardiac rhabdomyomas are benign muscle tumors and are detected prenatally with the use of sonography. They can occur sporadically, with congenital heart abnormalities, or with genetic disorders. They are most often associated with tuberous sclerosis complex (TSC), a genetic disorder, in approximately 60% to 96% of cases.1,2 Most rhabdomyomas spontaneously regress within the first few years of life. An intervention is only necessary when the rhabdomyomas obstruct normal heart function.
Case Report
A 31-year-old woman, G7 T4 P0 A2 L4, was referred to the maternal fetal medicine clinic in her late third trimester of pregnancy due to a positive hepatitis B surface antigen (HBsAg) test with elevated viral load detected earlier in her pregnancy. The patient had previously undergone a fetal anatomy sonogram at 19 weeks’ gestation at an outside facility and no abnormalities were noted.
She presented for her complete morphology sonogram and maternal fetal medicine consult at 35 weeks, 2 days. The sonogram was performed with a Philips EPIQ 7 (Philips Healthcare, Andover, Massachusetts) ultrasound machine and a 5-1 MHz curvilinear array transducer. Sonographic evaluation revealed multiple echogenic masses within the fetal heart and mild polyhydramnios. All other fetal anatomy appeared unremarkable. Upon discovery of the cardiac masses, a fetal echocardiogram was performed. The masses appeared to be intramyocardial, involving the interventricular septum and both the left and right ventricles (Figures 1 and 2). The largest mass was spheroid in shape and located in the apex of the left ventricle measuring 2.68 × 2.34 × 2.34 cm (Figure 3). Color Doppler evaluation of the masses demonstrated no blood flow within the cardiac lesions (Figure 4). The remainder of the fetal echocardiogram was unremarkable, demonstrating normal biventricular systolic function and no outflow tract obstruction (Figure 5). A normal three-vessel view was obtained with pulmonary artery and aortic diameters measuring within normal limits. The findings were thought to be most consistent with cardiac rhabdomyomas, and a genetics consult was recommended.
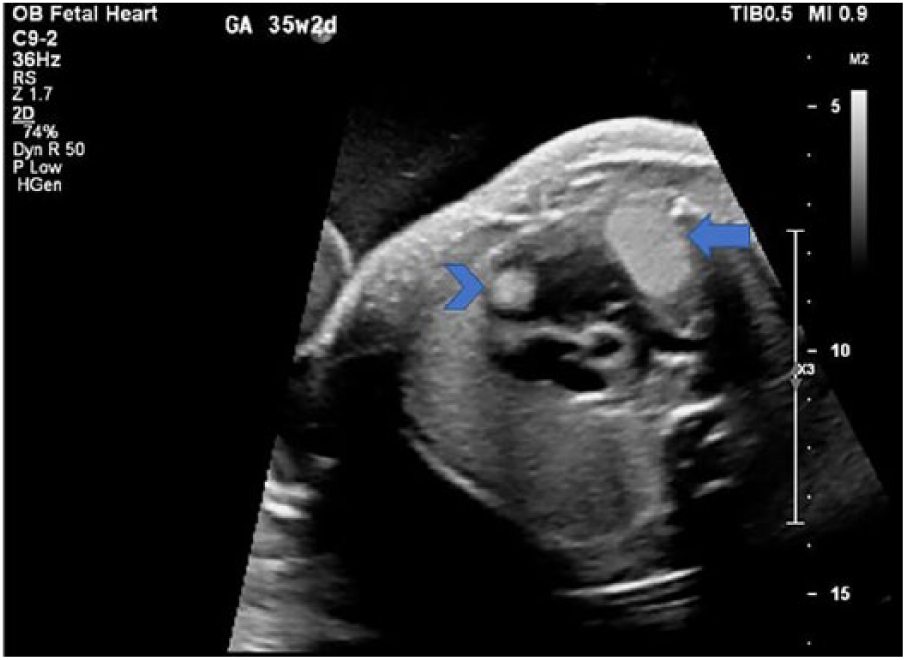
Fetal echocardiographic view showing two rhabdomyomas, the largest in the left ventricle (arrow) and a smaller one located in the right ventricle (arrowhead).
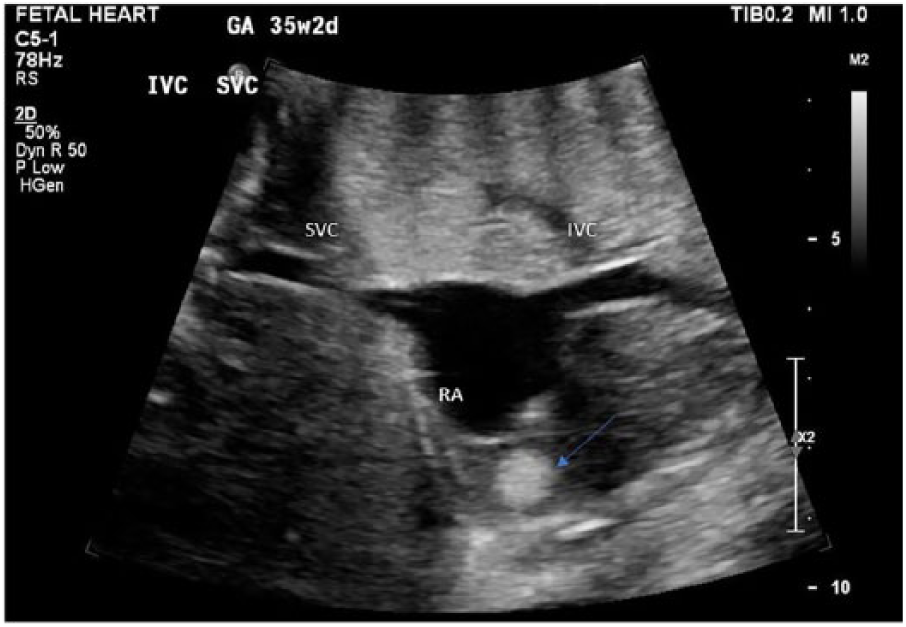
Fetal echocardiographic bicaval view showing a single rhabdomyoma in the right ventricular free wall (arrow).
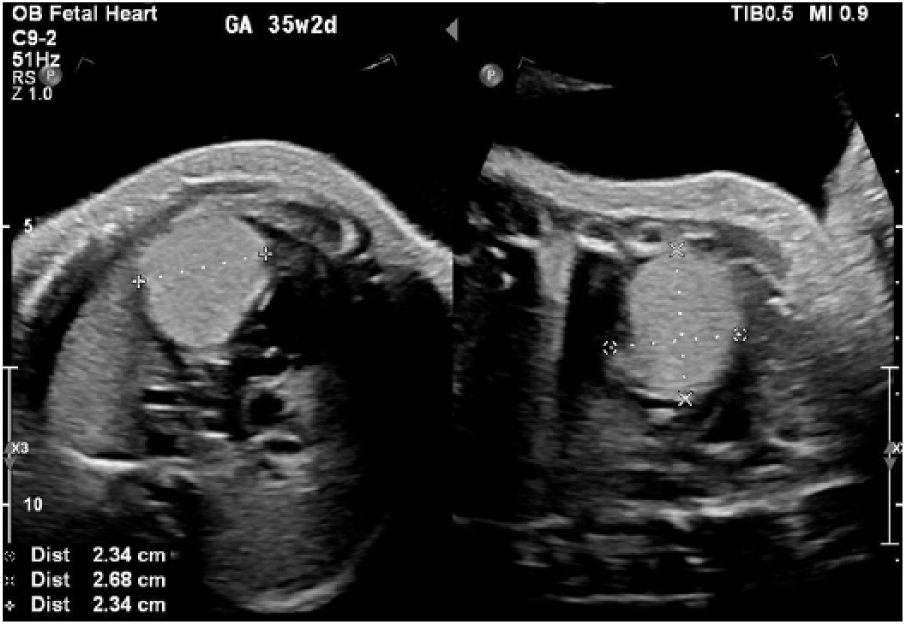
Fetal echocardiographic dual image showing the largest rhabdomyoma (calipers) located in the apex of the left ventricle.
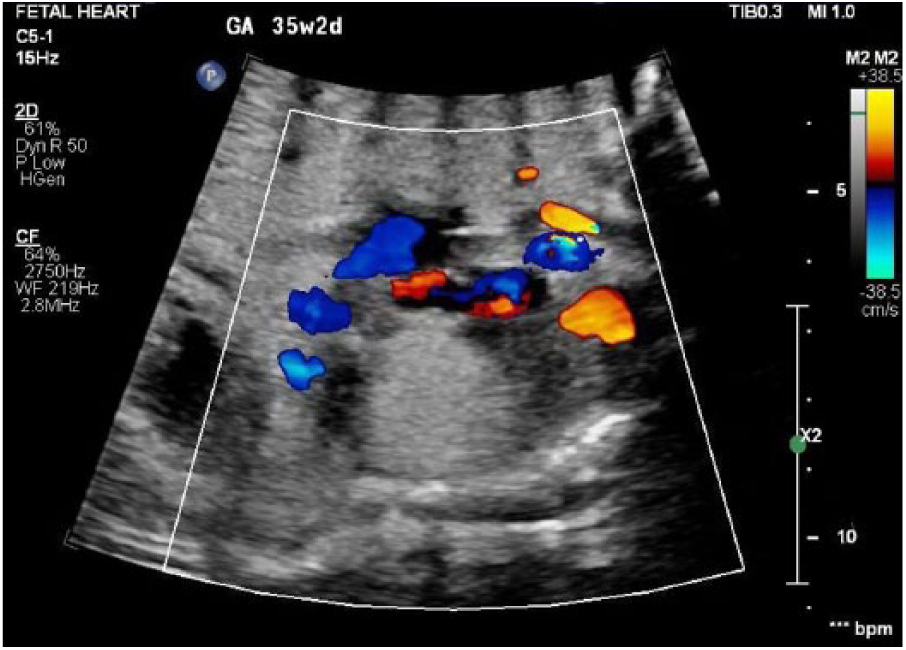
Color Doppler showing absence of blood flow within the largest rhabdomyoma.
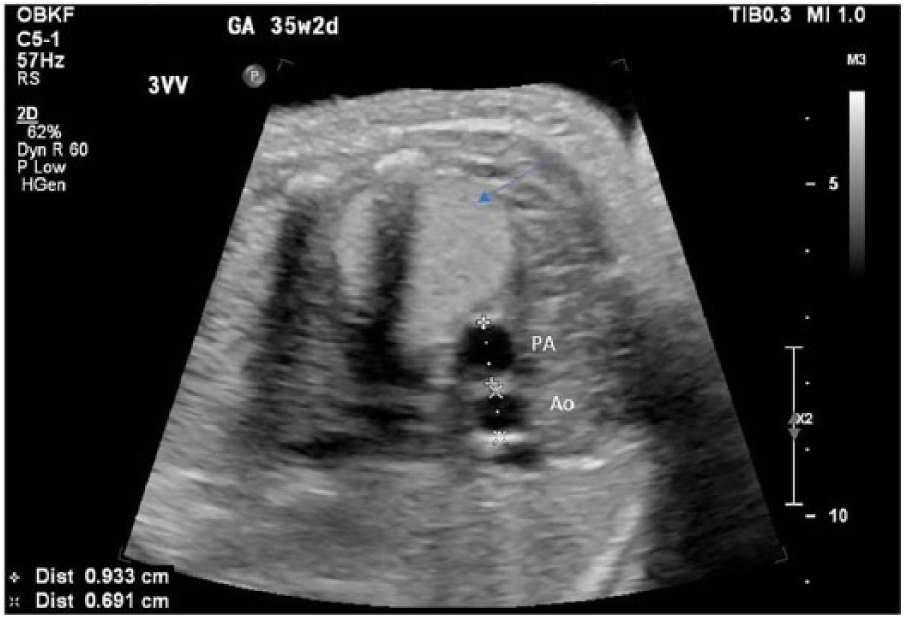
Fetal echocardiographic three-vessel view showing normal pulmonary artery (PA) and aortic (Ao) diameters. Also seen in this view is the largest rhabdomyoma in the left ventricle (arrow).
A genetics consult was performed following the sonogram, as findings of cardiac rhabdomyomas are typically associated with a diagnosis of TSC. Review of the maternal and paternal family history was negative for known birth defects; intellectual disability; epilepsy; behavioral disorders; tumors of the brain, eye, and kidney; skin lesions; lung disease; or any known genetic disorders. A family history of cholesterol, heart problems in the paternal grandfather, and cancer of unknown origin in both paternal grandparents was noted. The patient was found to have four phenotypically healthy children. Clinical evaluation and/or molecular genetic testing are essential to make a final diagnosis, as well as parental testing. In addition, rare cases or apparent nonpenetrance and germline mosaicism have been reported. The patient decided to postpone genetic testing of the fetus until after delivery.
The pregnancy was followed with weekly biophysical profile (BPP) sonograms. Each BPP sonogram received a score of 8/8 with reactive nonstress tests. Polyhydramnios was noted again each examination. The patient’s amniotic fluid index measured 30.29 cm, and the deepest vertical pocket measured 13.30 cm at 36 weeks.
The patient had an uncomplicated spontaneous vaginal delivery at 39 weeks, 6 days. The male neonate was admitted to the neonatal intensive care unit (NICU) 10 minutes following delivery and a hepatitis B vaccine and hepatitis B immunoglobin were administered shortly after admission.
A pediatric transthoracic echocardiogram was performed at two days of life. The echocardiogram was complicated by poor patient compliance. Multiple echogenic intracardiac masses were noted, involving the left ventricular and right ventricular free walls, interventricular septum, and left ventricular outflow tract. The largest mass appeared to be an oblong-shaped structure extending from the anterior interventricular septum into the right ventricular free wall and measured over 2.5 cm in the largest dimension. Another small echogenic mass was noted in the left ventricular outflow tract at the attachment of the anterior mitral valve that did not appear to obstruct antegrade flow. Multiple other small echogenic masses were seen throughout the myocardium and papillary muscles. Ventricular systolic function appeared normal and intracardiac flow appeared to be unimpeded.
A tuberous sclerosis panel/sequencing and deletion/duplication analysis of TSC1 and TSC2 was performed via cord blood. Results identified the individual was heterozygous for a pathogenic variant in the TSC2 gene. The result is consistent with the diagnosis of tuberous sclerosis complex.
The infant is otherwise doing well and has not been affected by the rhabdomyomas. The infant has shown appropriate weight gain and has had no issues feeding. Follow-up examination of the fetal heart revealed smaller masses compared to previous examinations. At this point, the infant does not need surgery. It is expected that the rhabdomyomas will regress spontaneously, and the only reasons for surgical intervention would be intractable arrhythmias or hemodynamic instability.
Discussion
Cardiac tumors are rarely detected in utero. Rhabdomyomas are the most common cardiac tumors diagnosed in both fetuses and infants, which accounts for 60% to 65% of fetal tumors. 3 The differential diagnoses may include but are not limited to myxomas, papillary fibroelastomas, fibromas, and teratomas. Rhabdomyomas are benign lesions that rarely invade surrounding structures and grow in utero due to the transmission of maternal estrogens to the fetus. They usually grow until 32 weeks’ gestation and then begin to gradually regress spontaneously. 4 They can be asymptomatic and create arrhythmias, as well as inflow and outflow obstructions, that can lead to cardiac death. 5 The symptoms are dependent on the size and location of the rhabdomyomas.
Primarily, multiple masses arise from the ventricular septum or free walls. Rhabdomyomas tend to regress within the first few years of life, and surgical resection is only needed when symptoms of obstruction are present. 6 Possible differential diagnoses for rhabdomyomas include fibromas and teratomas. A fibroma is an unlikely diagnosis because of the appearance and location of the masses. Fibromas usually present as a large single mass in the interventricular septum or the free wall of the left ventricle. Unlike rhabdomyomas, fibromas do not regress after birth. Teratomas are typically extracardiac masses, located near the aorta and pulmonary artery. 7 They are derived from two or three germinal layers of embryonic origin. They arise within the pericardium and are rarely found within the heart. Based on this patient’s findings, rhabdomyoma is the most likely diagnosis. 7
In utero, rhabdomyomas are detected during the second or third trimester by fetal echocardiography. 7 The most common sonographic appearance is homogeneous, nonvascular, echogenic, round masses, most often found in the left ventricle. 7 The mean diameter of a cardiac rhabdomyoma is 17 mm, but they can range in size and shape. Follow-up echocardiograms after birth are necessary to note the spontaneous regression of the rhabdomyomas and to ensure they are not causing more significant problems that can be detrimental to the infant’s health. 7
Rhabdomyomas are rarely an isolated finding. They are associated with TSC in 60% to 96% of cases.1,2 With the increasing practice, improvements of sonography, and the use of magnetic resonance imaging (MRI), these tumors are being diagnosed more frequently and accurately. The use of fetal echocardiography has increased the detection of rhabdomyomas in utero.8–12
The diagnosis of TSC can be made based on clinical findings and genetic testing; sonography alone cannot diagnose it. TSC is an autosomal dominant genetic disorder that affects approximately 1 in 6000 live births. Tumor formation is due to a mutation in the TSC1 or TSC2 gene. 13 TSC1 codes for the protein hamartin and TSC2 codes for tuberin. These two genes function to suppress the mammalian target of rapamycin, which is a serine-threonine kinase involved in cell proliferation and growth. 14 In familial cases, mutations in TSC1 and TSC2 are divided almost equally, whereas in sporadic cases, TSC2 mutations are more common. One-third of cases of TSC1 or TSC2 gene mutations occur as a result of a parent having the disorder. In the other two-thirds of cases, TSC1 or TSC2 pathogenic variants present as de novo mutations. 15 The severity of TSC can vary greatly among individuals who are affected even in the same family. Penetrance of TSC is assessed at 100% with inter- and intrafamilial variability in clinical features, which might present as negative family history. This may be due to family members who have previously escaped diagnosis because of a milder phenotype. 15 If one parent is affected or has the known familial pathogenic variant, the risk to the siblings is 50%. In the setting of a proband with a pathogenic variant and unaffected parents, the risk to the siblings being affected with tuberous sclerosis is 1% to 2%, due to germline mosaicism. Each child of an affected parent with TSC has a 50% chance of inheriting the pathogenic variant. 15
Benign tumors of TSC can manifest in the brain, heart, skin, eyes, kidneys, lungs, and liver. Most patients with TSC have at least one or more skin lesions that help characterize the disorder.16,17 The main cardiovascular characteristic of TSC is a rhabdomyoma.
Because of the spontaneous regression after birth, typically, no treatment is necessary. If they do produce symptoms, they are most likely to occur during the neonatal and infancy periods. 18 If the tumors increase in size following birth, there is a chance of obstruction of inflow and outflow in the heart, which can be fatal. In the cases of obstruction, surgical resection is needed. The use of everolimus, a drug therapy that targets the mammalian target of rapamycin pathway to increase the regression rate of the rhabdomyomas, has been found to be effective when they do not spontaneously regress. 19
The prognosis for TSC is highly variable and can have a range of phenotypic outcomes. If only cardiac rhabdomyomas are observed, the prognosis is good. Malignancy from cardiac rhabdomyomas has not been reported. In rare occasions, coarctation of the aorta, obstruction of major arteries, or an aortic aneurysm may occur. 20
TSC is most problematic in patients when brain manifestations are present. Brain lesions of TSC are primarily associated with developmental delay and epilepsy. 7 The lesions can be in the form of cortical tubers, subependymal nodules, subependymal giant cell tumors, or white matter heteropia.21,22 The lesions can be imaged with either MRI or sonography. They can appear as echogenic masses. However, the primary diagnosis of TSC is based on cardiac rhabdomyomas. 21 Renal lesions from TSC can develop as angiomyolipomas. Angiomyolipomas present as echogenic lesions in the kidney. These patients are at risk for developing chronic kidney disease. 20
Conclusion
Rhabdomyomas are most commonly associated with TSC. Cardiac rhabdomyomas usually regress within the first few years of life. In this case, tumors were only found in the heart and not in the brain or kidneys, and therefore the prognosis was good. 20 Detection of rhabdomyomas during the prenatal period with sonography is important for the well-being of the fetus. With early detection, the fetus can be further examined for other abnormalities. Delivery plans, proper postnatal care, and testing can be prepared if the rhabdomyomas are detected in utero.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.