
Abstract
Antley-Bixler syndrome (ABS)—consisting of ABS type 1 (skeletal only) and ABS type 2, associated with cytochrome 450 mutations and P450 oxidoreductase deficiency (PORD-ABS)—is a heterogeneous syndrome with a constellation of skeletal deformation findings that classically include skull, facial, and appendicular defects. The hallmarks of ABS type 1 include craniosynostosis, midface hypoplasia, radiohumeral/radioulnar synostosis, arachnodactyly, bowing of the femur, congenital fractures, and choanal atresia. ABS has historically been identified at newborn or childhood examination using radiographic, computed tomography, or stillbirth autopsy. However, in recent years, antenatal ultrasonography has identified craniosynostotic conditions in utero. We present a case of PORD with the ABS phenotype diagnosed by sonography in utero at an early gestation (13–16 weeks), which included mutations in the POR gene. Sonographers, sonologists, and perinatologists aware of the diversity of this rare condition will be well prepared to raise the suspicion of Antley-Bixler PORD-type syndrome.
Keywords
Antley-Bixler syndrome (ABS) is an exceptionally rare congenital disorder characterized by multiple musculoskeletal, craniofacial, and urogenital anomalies and a female predominance. 1 ABS was first reported by Antley and Bixler in 1975. 2 Since then, more than 50 cases have been reported in the world literature, 3 of which few cases were diagnosed prenatally.4–13 A case of Antley-Bixler syndrome (P450 oxidoreductase deficiency [PORD]–ABS phenotype, ABS type 2) is presented in which its associated anomalies of characteristic radiohumeral synostosis, femoral bowing, clubfoot, and facial features and abnormalities enabled a prenatal diagnosis of this rare entity to be made.
Case Report
A thirty-year-old woman, gravida 2, para 0010, was referred to our affiliated hospital at 22 weeks of gestation because of vaginal bleeding. The patient had an unremarkable family and medical history, and her previous pregnancy was remarkable for an elective abortion due to unspecified skeletal dysplasia. She denied any consanguinity with her husband and history of teratogenic exposure or medications.
Since the patient’s previous pregnancy was complicated by camptomelic dysplasia, extensive sonographic surveillance was conducted using a 6-MHz curvilinear array transducer (GE Voluson 720; GE Healthcare, Piscataway, New Jersey). The first sonogram showed an approximately 13-week singleton fetus consistent with the patient’s stated last menstrual period. A nuchal translucency evaluation showed a normal 1.9-mm thickness (Figure 1). Fetal anatomy surveillance, especially the fetal limbs, was indeterminate because of the early fetal gestational age.
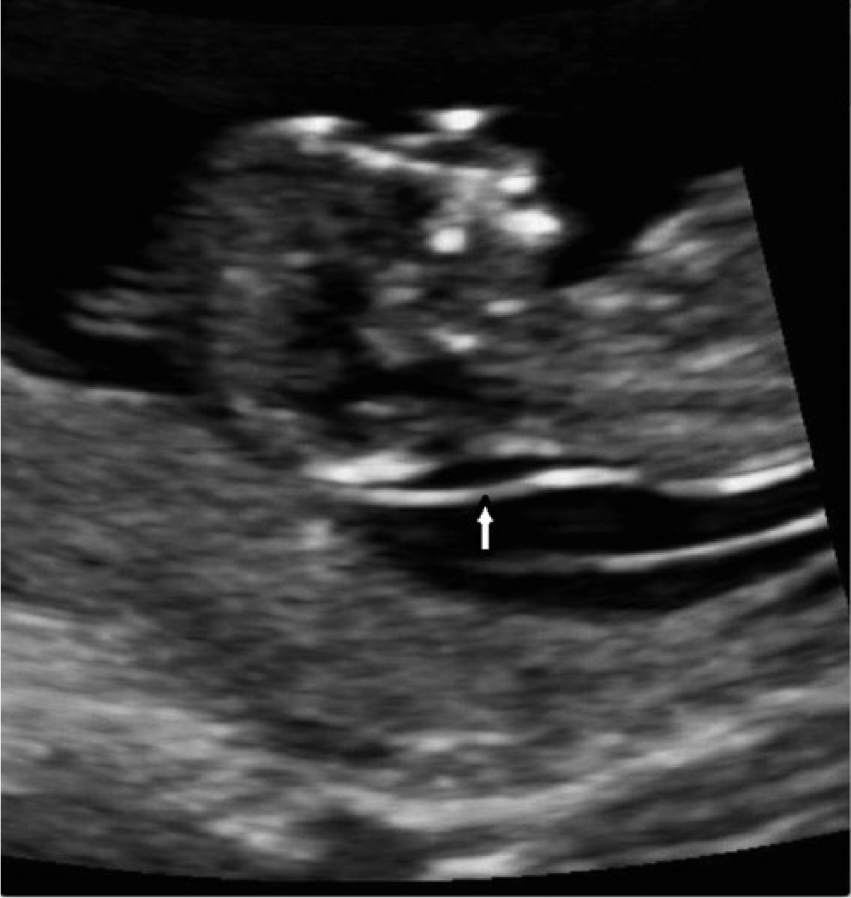
A sagittal view of the fetal neck demonstrates a normal nuchal translucency of 1.9 mm (arrow).
A recommended sonographic follow-up examination was scheduled at 22 weeks’ gestation. However, when unexpected vaginal bleeding occurred, the patient returned to our affiliated facility at 15 weeks’ gestation. At this time, an extensive sonographic examination of the fetus revealed bilateral femoral midshaft bowing (Figure 2A,B), clubbing with internal rotation of feet (Figure 3), and frontal bone flattening or “keel-shaped” configuration of trigonocephaly with small cisterna magna (Figure 4A,B). Because of the presence of these anomalies, a repeat sonographic examination was done one week later that confirmed the previous findings and further revealed a radiohumeral synostosis (Figure 5) as well as micrognathia and prominent nose (Figure 6). The differential diagnosis for these sonographic findings included trisomy 18, Jacobsen syndrome, camptomelic dysplasia, Pena-Shokeir syndrome, ABS, Apert syndrome, Muenke syndrome, Jackson-Weiss syndrome, Beare-Stevenson cutis gyrata, Crouzon syndrome, and Pfeiffer syndrome.
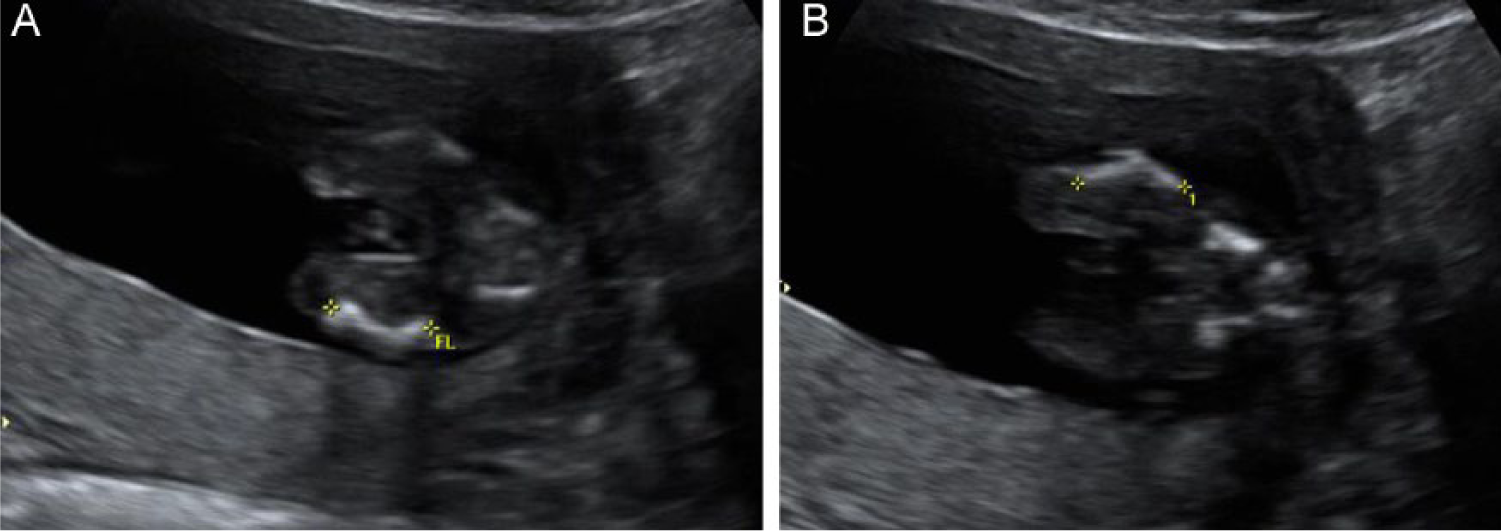
Sagittal views of the (A) left thigh and (B) right thigh demonstrate femoral shortening and midshaft bowing.
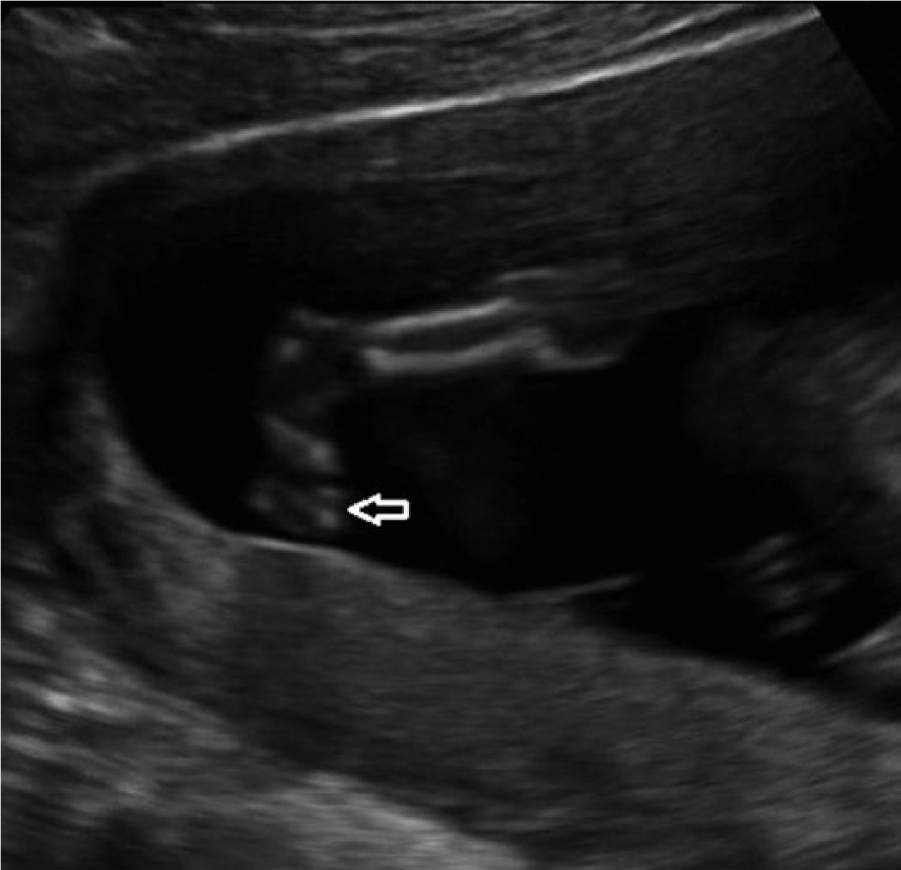
A 2D sagittal image of the right lower extremity shows mild clubbing with internal rotation of foot (arrow).
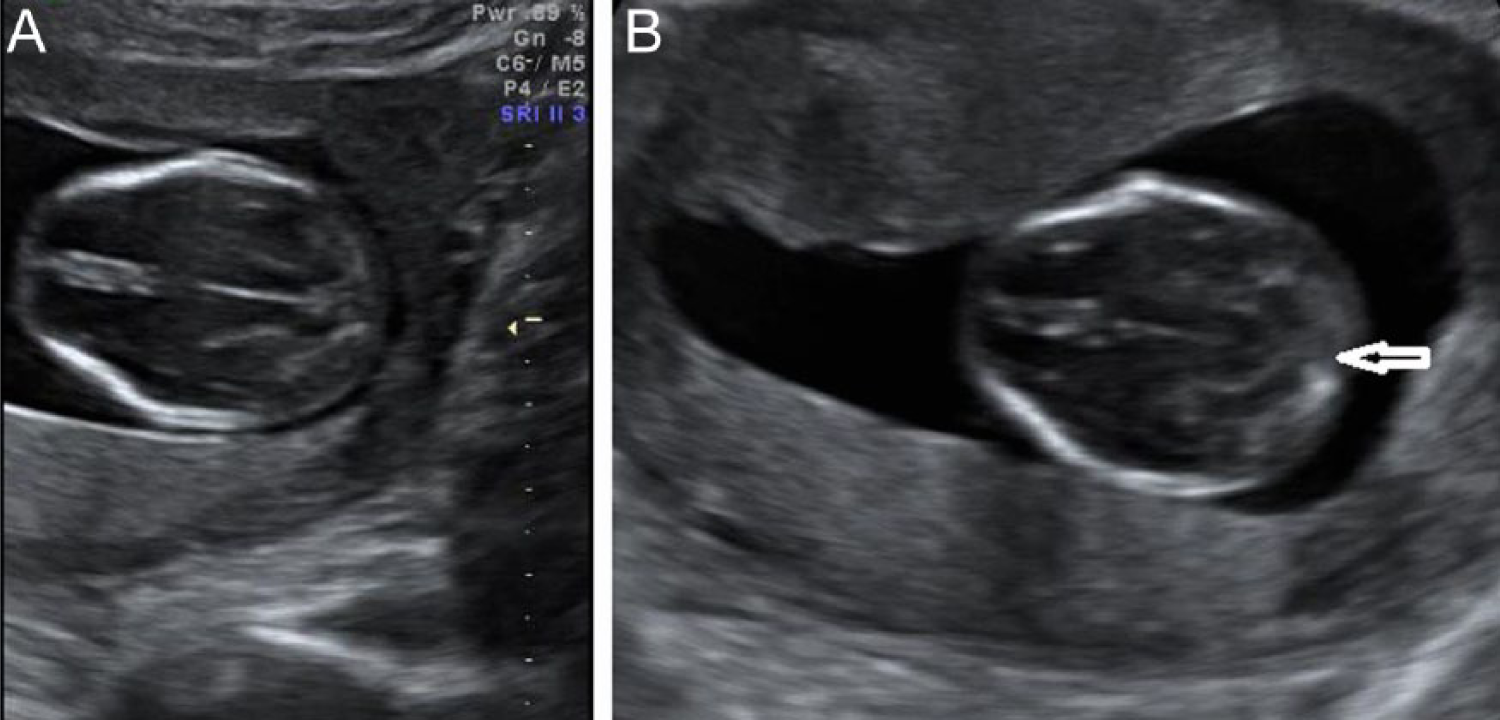
Transverse images of the fetal skull demonstrate (A) a “keel-shaped” configuration of trigonocephaly and (B) a small cisterna magna (arrow).
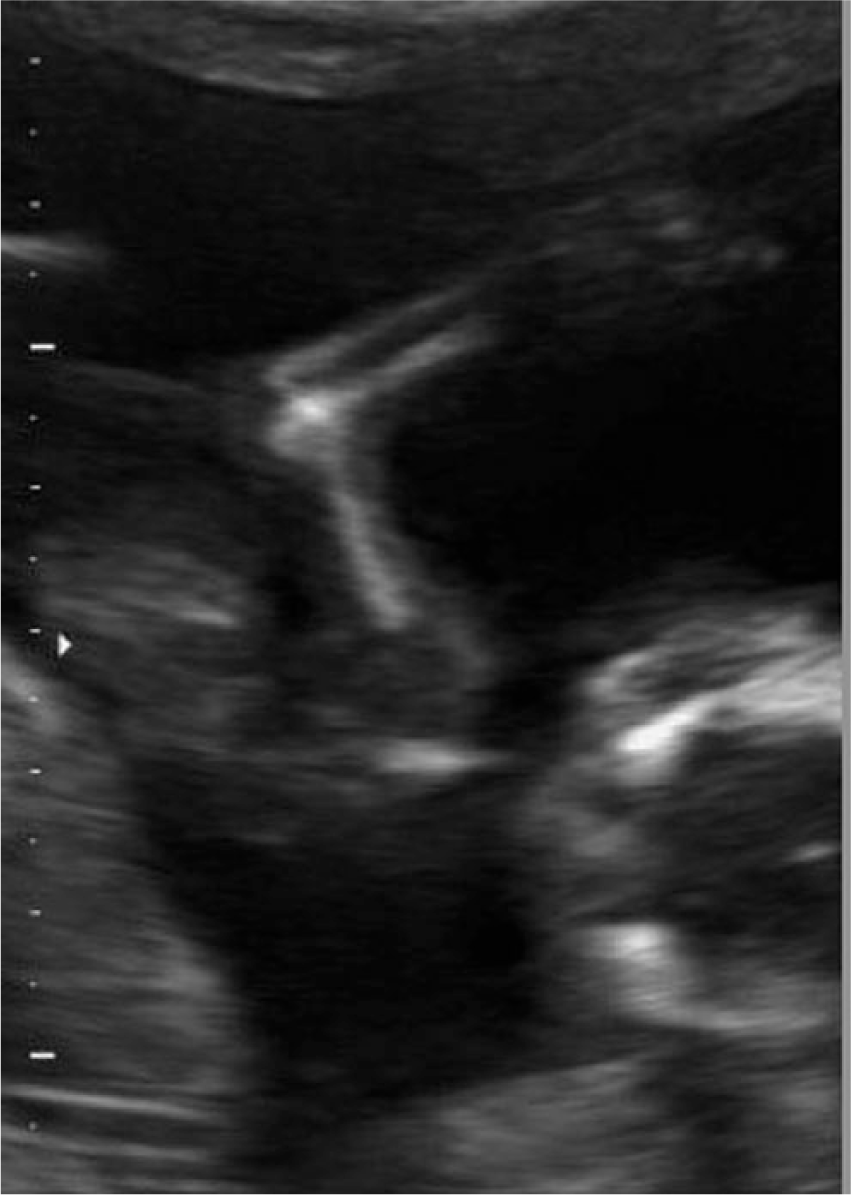
A sagittal view of the right upper extremity demonstrates a radiohumeral synostosis.
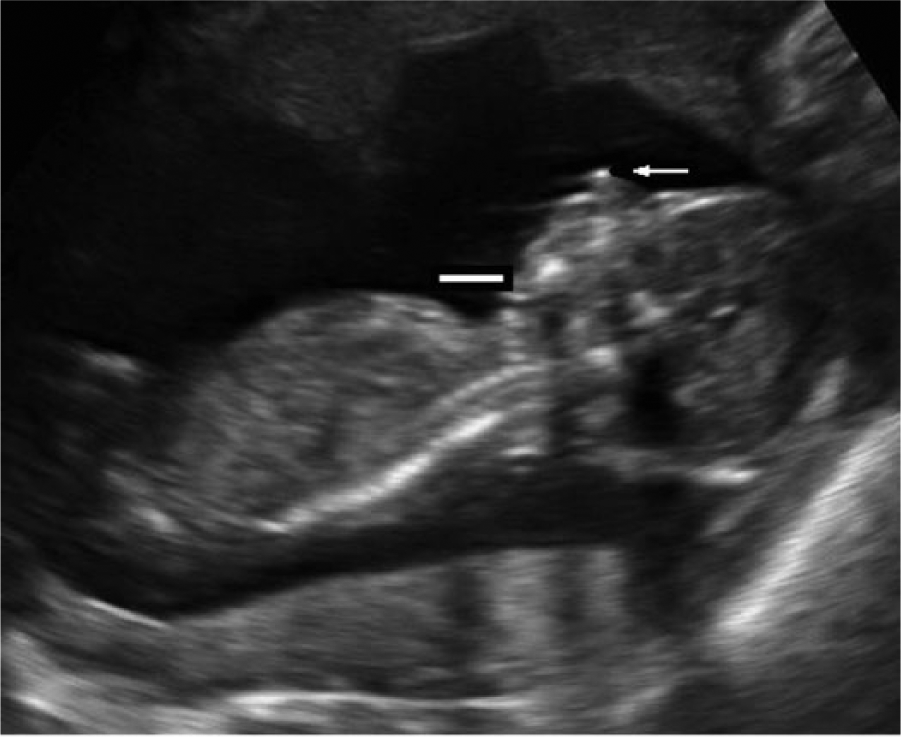
A fetal profile sonogram demonstrates micrognathia (solid line) and prominent nose (arrow).
Based on the obstetric history and the sonographic findings of craniosynostosis, radiohumeral synostosis, micrognathia, femoral shortening and bowing, and equinovarous talipes, a prenatal diagnosis of ABS was suspected. An amniocentesis showed a normal karyotype of 46, XX. A microarray chromosomal analysis on the cells from the amniocentesis was done that was negative for camptomelic dysplasia (absent SOX9 mutation). A second-trimester maternal serum screening was conducted and showed extremely low unconjugated serum estriol (UE3) levels. A cytochrome P450 oxidoreductase (POR) analysis was performed. Two disease-causing mutations (A287P missense and Q609X nonsense) were identified in the POR gene. These mutations were consistent with ABS and confirmed the prenatal suspicion. After termination of pregnancy, the prenatal diagnosis of ABS was further confirmed by autopsy, with the identification of prenatal sonographic findings.
Discussion
ABS is a dichotomous syndrome, consisting of a classic grouping of anomalies first noted in 1975. 2 The exact underlying cause of ABS remains unclear. Current thought views ABS as two distinct entities: (1) skeletal-only ABS (ABS type 1), inherited in an autosomal dominant fashion, caused by heterozygous mutations in the fibroblast growth factor receptor (FGFR) family, and thus related to other FGFR mutation syndromes such as Pfeiffer, Apert, Jackson-Weiss, and Crouzon, and (2) PORD (ABS type 2), inherited in an autosomal recessive fashion.1,14–18
ABS type 1 is exceedingly rare, with a less than 1 in 1 million live-birth occurrence rate. Much of the published research has involved families with more than one affected member; consanguinity increases the chance of multiple affected family members.6,19,20 The more recently identified ABS type 2 is caused by defects in cholesterol synthesis caused by mutation in one or both of the alleles for cytochrome PORD. This enzyme is critical in the production of steroids and also involved in skeletal formation. In addition, P450 oxidoreductase is involved in liver metabolism, activation, and detoxification of many drugs. Currently, there are about 25 mutations of the POR gene described. 21
In the case presented, two mutations in the POR gene were identified. One is a G>C nucleotide substitution in exon 9, resulting in the replacement of an alanine codon (GCT) with a proline codon (CCT) at amino acid position 287, denoted c.859 G>C at the complementary DNA (cDNA) level or p.Ala287Pro (A287P) at the protein level, and the C>T nucleotide substitution in exon 15, resulting in the replacement of a glutamine codon (CAG) with a stop codon (TAG) at amino acid position 609, denoted c.1825 C>T at the cDNA level or p.Gln609Stop (Q609X) at the protein level. The A287P missense mutation in the POR gene is a common mutation that has been identified in patients with ABS. 22 To our knowledge, the Q609X nonsense mutation has not been previously reported, and this warrants future investigation of its impact on ABS. There is a very wide spectrum of phenotype and clinical conditions in PORD, including extremely mild congenital adrenal hyperplasia and very subtle craniofacial/skeletal abnormalities; more moderate manifestations, including ambiguous genitalia; and very severely affected fetuses that die in utero or at birth.21–23 In addition, high-dose fluconazole intake by pregnant mothers during early pregnancy has been reported as a possible etiology of ABS.19,24–26
Both types of ABS share primarily skeletal malformations, which are characterized by craniosynostosis,2,5–7,20,27–29 brachycephaly,7,12,13,28–30 choanal atresia or stenosis,5–7,20,27,28 frontal bossing,5–7,20,27–29 anteverted nostrils and proptosis of the eyes,5,7,27,29 and midface hypoplasia.5,7,17,27,29 In addition, radiohumeral and radioulnar synostosis, femoral bowing, femoral fractures, narrow rib cage and pelvis, camptodactyly, arachnodactyly with bulbous fingertips, clubfeet, multiple contractures, and ambiguous genitalia have also been reported.2,5–7,20,27–29,31 Other malformations such cardiac defects, renal duplication, large clitoris, imperforate anus, cryptorchidism, fused labia majora, and hydrocephalus have been described.2,6,20,32,33
ABS has classically been identified at newborn or childhood examination using radiography, computed tomography, magnetic resonance imaging, or stillbirth autopsy. However, in recent years, ultrasonography has enhanced the prenatal diagnosis of ABS as it is able to document many of the clinical features associated with this syndrome prenatally.4–6,9,12,34,35
Many of the phenotypic features that have been described and reported prenatally include femoral shortening and bowing,6,11,12 radiohumeral synostosis,5,6 craniosynostosis and trigonocephaly,4–7,12,36 micrognathia, depressed nasal bridge, facial hypoplasia, abnormal curvature of the feet, and immobility and fixed flexion of the joints of the elbows.4,5,12
Other prenatal sonographic features such as hypertelorism, palpebral edema, exophthalmia, polyhydramnios, choanal atresia (demonstrated by lack of communication between the nostrils and nasopharynx by Doppler evaluation), lemon-shaped skull, horseshoe kidneys, renal agenesis, bowing of the ulna, and absence of nasal bones have also been reported.4–6,12,32 In addition to the clinical and sonographic features, ABS/PORD has been associated with extremely low (or undetectable) maternal serum estriol levels, as was evident in this case. Consequently, maternal serum estriol levels may strengthen the prenatal diagnosis of ABS.8,37
Although sonography has improved the prenatal diagnosis of ABS, in some cases, a definitive diagnosis can be uncertain because of the numerous sonographic findings shared with other skeletal dysplasia disorders such as trisomy 18, Jacobsen syndrome, camptomelic dysplasia, Pena-Shokeir syndrome, Apert syndrome, Muenke syndrome, Jackson-Weiss syndrome, Beare-Stevenson cutis gyrata, Crouzon syndrome, and Pfeiffer syndrome. In this case, the prenatal suspicion of ABS was strengthened by the presence of femoral shortening and bowing, radiohumeral synostosis, micrognathia, trigonocephaly, craniosynostosis, and equinovarous talipes. Current thought in the field holds that craniosynotosis, radiohumeral synostosis, and femoral bowing with extremely low maternal estriols levels are indicative of this syndrome, which correlates with the findings in this case.8,37,38
ABS carries implications for brain development. The skull malformation can lead to mental retardation due to poor brain development. 19 In addition, airway malformation such as choanal atresia or stenosis may lead to respiratory distress, which causes more than 90% of the deaths in ABS-affected neonates. 4 However, with prompt intervention, including tracheostomy and alleviation of the craniosynostosis through infant surgery, the prognosis can be “quite satisfactory development.”1,5,6,9,34 Regarding synostosis specifically, many other authors believe the prognosis of resecting or correcting it is very poor, and surgical interventions should be avoided due to recurrence.7,17,27 In general, active physical therapy has been reported to be effective in maintaining and improving the range of movement in patients with ABS.1,11 Overall, the treatment of ABS malformations is usually tailored to each patient and to the severity of the anomaly.
Conclusion
Sonographic imaging technology has enabled sonographers, sonologists, and perinatologists to directly visualize and identify various fetal abnormalities. ABS should be suspected at sonography when femoral shortening and bowing, radiohumeral synostosis, micrognathia, trigonocephaly, craniosynostosis, and equinovarous talipes are identified. These sonographic features, along with low maternal serum estriol levels and the presence of mutations of the cytochrome P450 oxidoreductase gene, support the diagnosis of this rare genetic syndrome. The prenatal diagnosis of this syndrome may provide a critical data point for parents to decide on continuation of pregnancy; on preparations for early medical support, surgical intervention, and treatment; and on the provision of adequate counseling about the long-term prognosis of the syndrome.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.