
Abstract
Hypomyelination leukodystrophies (HLDs) are a group of rare genetic disorders that impair myelin formation in the central nervous system (CNS), leading to developmental delays and neurological symptoms. HLD type 11 (HLD11) is caused by mutations in the POLR1C gene, manifesting with a 4H phenotype (hypomyelination, hypodontia, and hypogonadotropic hypogonadism) and associated neurodevelopmental delays. Here, we report a case of a male patient diagnosed with HLD11 who presented with developmental delay, hypotonia, and cerebellar atrophy. Whole exome sequencing identified a homozygous likely pathogenic POLR1C variant. Notably, the patient developed diabetes, an association not previously documented in HLD11. Despite intensive care, he ultimately passed away due to complications at 3.5 years of age. This report highlights the importance of thorough endocrinological evaluation in HLD patients and underscores the need for further research to understand the full spectrum of HLD11 manifestations.
Introduction
Hypomyelination leukodystrophies (HLDs) are a heterogeneous group of inherited white matter disorders defined by substantial and permanent deficits in central nervous system (CNS) myelin deposition. 1 With a prevalence of 0.78 per 100 000 people in individuals aged 0 to 19 years old, 2 This disease causes developmental delay and spasticity which was initially discovered by Pelizaeus. 3 Twenty-five years later, Merzbacher 4 provided more details about the neuropathology of twelve affected proband relatives. Based on the defect in a specific gene involved in myelination the classification for HLDs is established. 5 HLD type 11 (HLD11) is caused by a homozygous or compound heterozygous mutation in the POLR1C gene. Patients with HLD11 manifest a phenotype similar to HLD7 and HLD8 (4H phenotype). 5 4H leukodystrophy is a hypomyelinating leukodystrophy caused by biallelic pathogenic mutations in POLR3A, POLR3B, POLR1C, and POLR3K, all of which encode subunits of the POLR3 complex. It is associated with several distinctive neurological (usually cerebellar manifestations) and non-neurological features, including myopia and endocrine abnormalities such as growth hormone (GH) deficiency and short stature. The condition primarily includes hypomyelination, hypodontia, and hypogonadotropic hypogonadism, which are associated with a distinct MRI phenotype. 1 While there is abundant literature about HLDs, our literature review reveals a lack of specific literature available for HLD11 patients due to the rare cases. Here we present a patient with HLD11 with 4H phenotype accompanied by diabetes that hasn’t been defined previously in the literature. Whether this is coincidental or the phenotypic expansion is unknown.
Case Presentation
A male patient was observed from infancy to 3.5 years old. He was delivered via cesarean section due to macrosomia, with a birth weight of 4.03 kg, born to parents in a first-degree consanguineous marriage. The delivery was uneventful, resulting in a seemingly healthy infant.
At 10 months of age, the mother reported concerns about neurodevelopmental delays, including hypotonia, an inability to sit or crawl without support, and a lack of laughter and babbling. She noted that there was no similar history of developmental delay among his siblings. Despite these delays, some developmental milestones were met: spontaneous smiling by 4 months, recognition of faces and response to his name by 6 months, and object manipulation by 8 months.
A brain MRI conducted at 1 year of age showed generalized bilateral abnormal white matter signal intensity in both cerebral hemispheres and posterior cranial fossa structures, characterized by low signal intensity on T1-weighted (T1W) images and high signal intensity on T2-weighted (T2W) images. This was associated with a lack of myelinated structures that are typically present from birth, suggestive of inherited leukodystrophy (Figure 1). Additionally, moderate cerebellar atrophy was noted, with the brainstem size preserved (Figure 2).
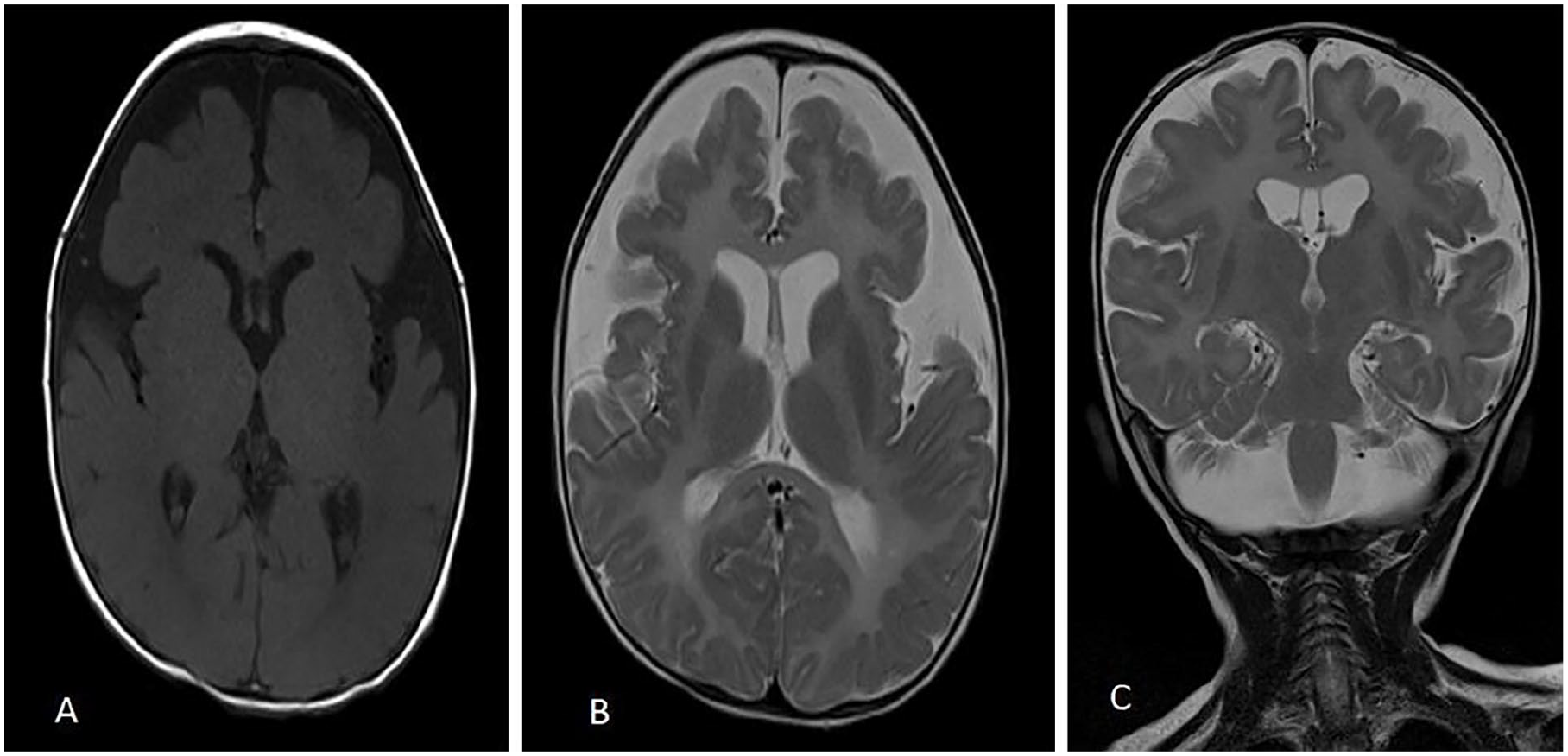
The axial T1-weighted (T1W) (A), axial T2-weighted (T2W) (B), and coronal T2-weighted (C) sequences of brain MRI demonstrate diffuse low T1W signal intensity and high T2W signal intensity within the white matter. These findings are consistent with hypomyelinated white matter.

Mid-sagittal T2W image, shows cerebellar atrophy.
At 1 year and 2 months of age, the patient presented to the hospital with symptoms of cough, fever, hypotonia, polyuria, and polydipsia. Physical examination indicated signs of illness, lethargy, dehydration, distress, and deep, rapid breathing. The Glasgow Coma Scale (GCS) score was 12/15, with the patient demonstrating an open-eye response to verbal stimuli, localized pain, and an irritable cry. Growth parameters were as follows: fronto-occipital head circumference was 45.5 cm (around the 50th percentile), weight was 7.3 kg (below the 5th percentile), and length was 77 cm (approximately the 10th percentile). Complete head lag was observed, with muscle power assessed at 3/5, and a positive Babinski reflex was noted.
Laboratory findings revealed hypoglycemia, ketosis, high anion gap metabolic acidosis, low C-peptide levels, elevated random blood glucose (RBS) of 653 mg/dL, and a high HbA1c of 7.9%. The initial suspicion was Type 1 Diabetes Mellitus (DM1) presenting with diabetic ketoacidosis (DKA), and the patient was managed in the pediatric intensive care unit (PICU). Upon discharge, the patient was prescribed a total daily insulin dose of 4 units, consisting of long-acting insulin (insulin glargine): 1 unit at 8 PM and 1 unit at 8 AM, and short-acting insulin (Actrapid): 2 units per day (divided as needed based on meal times).
Within the next month, tests for islet cell antibodies and anti-glutamic acid decarboxylase (anti-GAD) antibodies were performed, and both returned negative. This led to further investigation into the type of diabetes, including genetic testing for monogenic causes, which yielded non-informative results. Given the circumstances, a functional genomics study was considered to clarify the type of diabetes further. However, this test was unavailable in our facility, and the patient was unable to seek it elsewhere. As a result, the possibility of seronegative DM1 remains.
A follow-up brain MRI 1 year later showed no improvement in myelination (Figure 3), prompting further investigation. Subsequently, the patient’s whole exome sequencing (WES) revealed a significant homozygous variant in the POLR1C gene, specifically c.205G>T, resulting in the amino acid change p.Asp69Tyr. This variant is located at genomic position 6:43487134 in the transcript NM_203290.4. The variant is classified as “likely pathogenic” according to ACMG guidelines. This genetic alteration aligns with the clinical diagnosis of HLD11.

The axial T1-weighted (T1W) (A), axial T2-weighted (T2W) (B), and sagittal T2-weighted (C) sequences of brain MRI show the same signal intensity of the previous image (Figure 1), which is presumed to be myelinated (reverse signal). Note also cerebral atrophic changes.
At the age of three, the patient was admitted for a comprehensive evaluation following a single seizure episode lasting less than 1 minute, which resolved spontaneously and did not recur. Ophthalmic examination revealed poor eye contact and horizontal nystagmus; however, there were no prior ophthalmology consultations or reports to determine if the patient had myopia. A neurological examination indicated long tract signs with exaggerated deep tendon reflexes. Growth parameters were as follows: fronto-occipital head circumference was 46 cm (at or below the 5th percentile), weight was 8 kg (at or below the 3rd percentile), and length was 80 cm (at or below the 3rd percentile). A dental examination showed multiple teeth with cavitation. Although teething of the upper incisors began at 1 year of age, the remaining deciduous teeth erupted much later and were fully present by the age of three. According to the mother, these teeth subsequently began to show abnormal changes.
The patient’s final admission at the age of 3.5 years was due to a five-day fever. A comprehensive history and detailed physical examination are all unremarkable. Laboratory studies include complete blood count (CBC) with differential, arterial blood gas (ABG), electrolytes, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), serum transaminases, urinalysis, urine culture, and blood cultures, all of which are normal. Imaging options, including chest radiography and echocardiography, were also normal. So, an initial diagnosis was made as a fever of unknown origin, and the rest of the tests were requested, which included antistreptolysin O (ASO), antinuclear antibody (ANA), rheumatoid factor (RF), and stool analysis. However, he exhibited distress, a decreased level of consciousness, oliguria, gross hematuria, and tachycardia before the tests were done. Despite efforts in the PICU, the patient developed cardiopulmonary arrest and, unfortunately, could not be resuscitated.
Discussion
HLD11, also known as POLR1C-related hypomyelinating leukodystrophy, is an autosomal recessive type of leukodystrophy characterized by delayed psychomotor development and other neurological features including intellectual disability, tremor, ataxia, spasticity, and cerebellar symptoms. Other non-neurological symptoms include myopia and dental abnormalities, and possibly hypogonadotropic hypogonadism associated with hypomyelination on MRI. 6 It is reported that this type is caused by homozygous or compound heterozygous mutations affecting the POLR1C gene located on chromosome 6p22. 1 This affected gene encodes a shared subunit between two RNA polymerases (I and III). Specific mutations in POLR1C that cause HLDs can decrease binding to POLR3 targets. This was discovered in a study conducted by Thiffault et al. 6
The diagnosis of HLDs was defined by both MRI and clinical features, in addition to genetic tests to identify the type based on the involved mutations that were determined. The diagnostic MRI findings include diffuse hypomyelination, thin corpus callosum, and cerebellar atrophy,5,6 and relative preservation of specific structures, namely the dentate nucleus, optic radiations, the anterolateral nucleus of the thalamus, globus pallidus, and corticospinal tracts at the level of the posterior limb of the internal capsule. 1 These display mild white matter T2W-hyperintensity with T1W-iso intensity or T1W-hyperintensity relative to the gray matter of the brain. 1 Persistent hypomyelination is important for distinguishing HLDs from delayed myelination in patients younger than 2 years old, as delayed myelination typically shows improvement in myelination on MRI images. 7
Endocrine abnormalities in patients with HLD11 are not fully understood. However, a recent study conducted in 2021 by Pelletier et al. 1 Involved 150 patients, with 56 having pathogenic variants in POLR3A, 81 in POLR3B, and 13 in POLR1C, the study described some endocrine features found in those patients. Findings in patients with the POLR1C gene included one patient with low growth hormone based on growth hormone stimulation testing. Two out of four patients had an elevation in prolactin levels. Among eight patients who had TSH levels tested, TSH was high in one of them, free thyroxine level was low in one, high in another, and normal in the rest. Our patient exhibited normal TSH and thyroxine levels. Additionally, hypogonadotropic hypogonadism could not be tested due to the patient’s young age.
The treatment of such patients remains palliative. However, with advancements in molecular diagnostic tools that identify many mutations, there are promising prospects for a cure with more specific targeted therapies. 8
Based on our literature review, two cases of siblings with the 4H syndrome (POLR3A-related disorder-HLD7 and POLR3B-related disorder-HLD8) were reported to have DM 1. 9 This study and our reported case emphasize the need for further research into endocrinological associations for patients with HLDs.
Conclusion
Our case report highlights an unusual association between HLD11 and diabetes, not previously documented in the literature. This novel finding emphasizes the need for comprehensive evaluation and management of patients with HLDs, considering potential additional medical complexities. Further research into such associations is crucial for improving diagnostic accuracy and patient outcomes in such cases.
Footnotes
Acknowledgements
The authors thank the Polytechnic Medical Students’ Research Association (PMRA) for their invaluable input and support throughout the research process. We also extend our gratitude to Hebron Governmental Hospital, Makassed Hospital, Al-Ahli Hospital, the Hebron branch of the Palestine Red Crescent Society, and Hadassah Medical Center for providing patient data.
Author Contributions
Q.A. contributed to the drafting, editing, and final revision of the article. D.A., K.J., L.A., and Y.R. were involved in the data collection and drafting of the article. M.S. contributed to the drafting process, while R.M. was responsible for editing and the final revision. All authors reviewed and approved the final manuscript.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Statement
Our institution does not require ethical approval for reporting individual cases or case series.
Patient Consent
Written informed consent was obtained from the patient’s family for the publication of this case report.