
Abstract
Extranodal Rosai–Dorfman disease (RDD) is a rare histiocytic disorder that typically presents with histiocyte proliferation in tissues outside of the lymph nodes, such as the central nervous system (CNS), posing significant diagnostic challenges. The pathogenesis of extranodal RDD remains unclear, though it is believed that genetic and immune factors play a role. In this study, we collected clinical, imaging, and histopathological data from three cases of extranodal RDD and analyzed formalin-fixed, paraffin-embedded tissue samples by using whole exome sequencing (WES) to identify mutations. Imaging findings showed extranodal RDD involvement in the CNS, chest wall, and right cheek, with histopathology confirming classical features of the disease. WES identified the rs368284573 mutation in the Phospholipase C Like 2 (PLCL2) gene across all cases. Experimental analysis of this mutation revealed a reduction in p-GSK-3β levels, which in turn inhibited GSK3β activity, leading to increased β-catenin accumulation, a key factor promoting cell proliferation. Knocking down the mutant PLCL2 gene restored p-GSK-3β levels, reduced β-catenin expression, and inhibited cell proliferation. These findings suggest that the PLCL2 mutation contributes to dysregulation of GSK3β/β-catenin signaling in extranodal RDD, providing new insights into the pathogenesis and potential therapeutic targets for this rare disorder.
Introduction
Rosai–Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is an uncommon non-Langerhans cell histiocytosis characterized by the proliferation of distinctive histiocytes within lymph nodes and various extranodal tissues. 1 The disease exhibits a slight male predominance and usually manifests in the first two decades of life, with nearly 80% of cases occurring before age 20. It has a higher prevalence among individuals of African descent, though cases have been reported worldwide across various ethnicities. 2
The concept of RDD was initially introduced by Destombes in 1965 and later acknowledged as a distinct clinical entity by Rosai and Dorfman in 1969. RDD typically manifests with painless cervical lymphadenopathy; however, extranodal involvement is frequently observed, affecting approximately 43% of cases.1,3 Extranodal RDD can affect various organs, including the skin, nasal cavity, soft tissues, and central nervous system (CNS), leading to a wide range of clinical and imaging manifestations. 4 This diversity often results in misdiagnosis or delayed diagnosis, which requires increased awareness and understanding of the disease.
The precise etiology of RDD remains partially unclear; however, recent research has suggested that it may result from a combination of genetic, immune, and infectious factors. Genetic studies have revealed mutations in key genes associated with the Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase (MAPK/ERK) signaling pathway, 5 such as Neuroblastoma RAS Viral Oncogene Homolog (NRAS), 6 Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS), 7 Mitogen-Activated Protein Kinase 1 (MAP2K1), 8 A-Raf Proto-Oncogene, Serine/Threonine Kinase (ARAF), 6 and Solute Carrier Family 29 Member 3 (SLC29A3), 9 supporting the hypothesis of genetic involvement in RDD’s pathogenesis. Immune dysregulation is also implicated, as the disease has been observed in association with autoimmune disorders, such as systemic lupus erythematosus and rheumatoid arthritis.10,11 Furthermore, there is evidence linking RDD to certain infections, with Epstein–Barr virus and human herpesvirus-6 being proposed as potential triggers.12,13 However, these infectious associations are inconsistent, and the mechanisms underlying their potential contribution to the development of RDD remain speculative. Collectively, these findings imply that RDD may not represent a singular, homogeneous entity but rather encompass a spectrum of disorders influenced by diverse pathogenic factors.
The emergence of whole exome sequencing (WES) has facilitated the discovery of genetic mutations associated with RDD, providing valuable insights into its pathogenesis and potential therapeutic targets. 14 However, the role of these mutations in the disease’s development and progression remains unclear. Therefore, this study aimed to utilize WES to analyze three cases of extranodal RDD from our hospital, detailing their clinical presentations, radiological features, and pathological findings. By identifying potential gene mutations in these cases, we aim to enhance the understanding of RDD’s pathogenesis and explore opportunities for potential targeted treatment strategies.
Results
Clinical data
The three patients, aged 31–69 years, included one male and two females, all presenting with extranodal RDD. Each case involved a different site, including the CNS, chest wall, and right cheek. Case 1 (CNS involvement) presented with symptoms of dizziness, nausea, and tinnitus. Case 2 (chest wall involvement) experienced chest pain. Case 3 (right cheek involvement) had a swelling and a painful lump in the cheek area. After the follow-up period, no recurrences were observed in any of the patients (Table 1).
Demographic and clinical characteristics of included patients
CNS, central nervous system; RFS, relapse-open-access survival.
Imaging findings
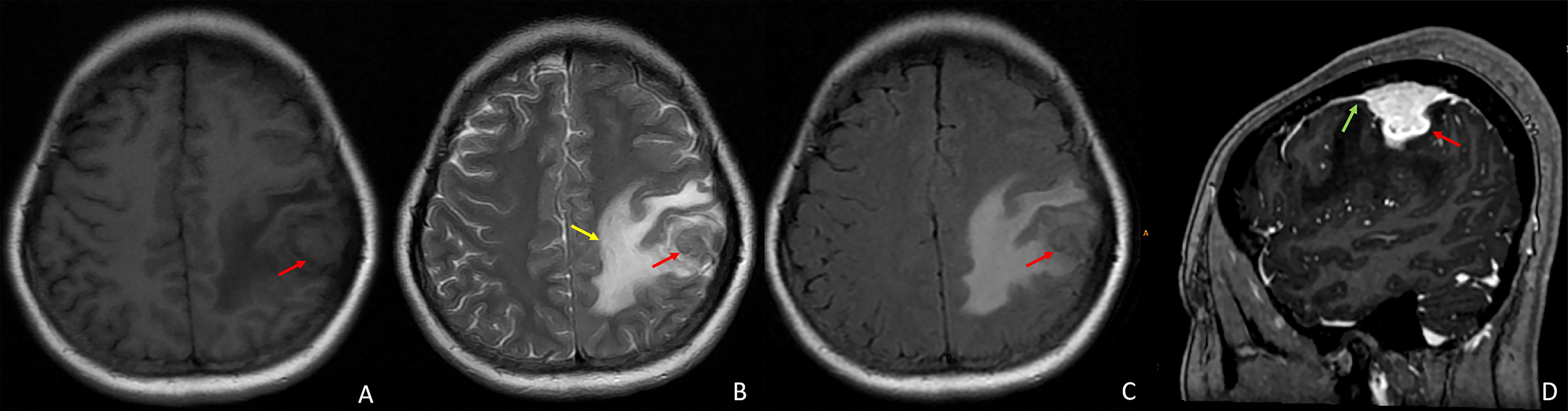
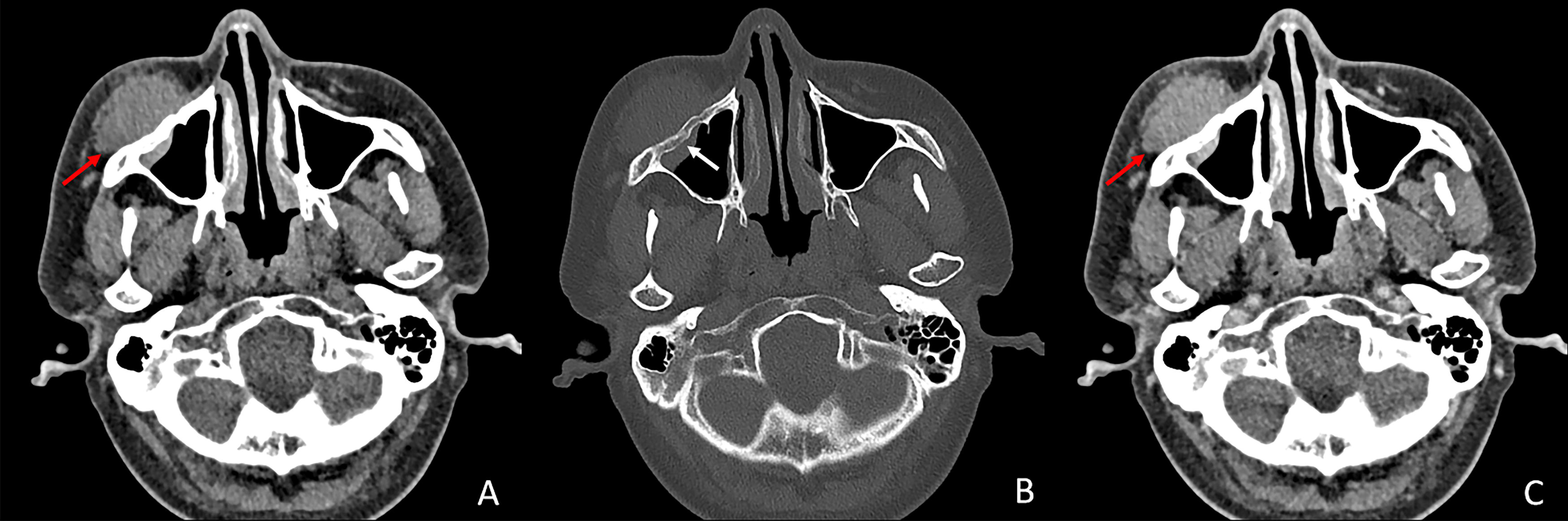
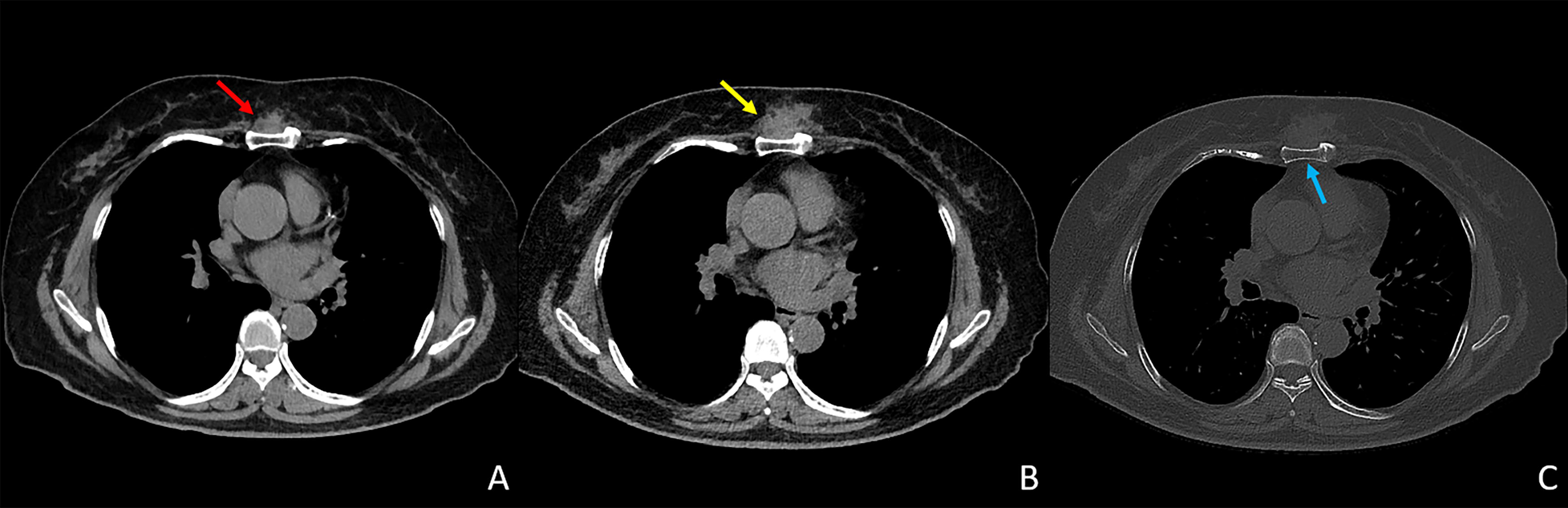
Computed tomography (CT) and magnetic resonance imaging (MRI) findings for the three cases are illustrated in Figures 1–3. In case 1, MRI of the left frontal lobe revealed the presence of a mass lesion. On T1-weighted MRI, the lesion exhibited slight hypointense, while on T2-weighted imaging, it displayed mild hyperintensity with surrounding patchy edema. Contrast-enhanced scanning showed heterogeneous enhancement of the lesion, with a distinctive dural tail enhancement at the periphery (Fig. 1). In case 2, a visible soft tissue mass is observed in the right cheek region, accompanied by thickening of the anterior wall of the right maxillary sinus. The enhanced scan reveals moderate and homogeneous enhancement of the lesion (Fig. 2). In case 3, a mass-like soft tissue opacity was observed anterior to the sternum. Over an 8-month follow-up period, there has been progressive enlargement of the lesion with no significant adjacent sternal abnormalities detected (Fig. 3).
Pathological findings
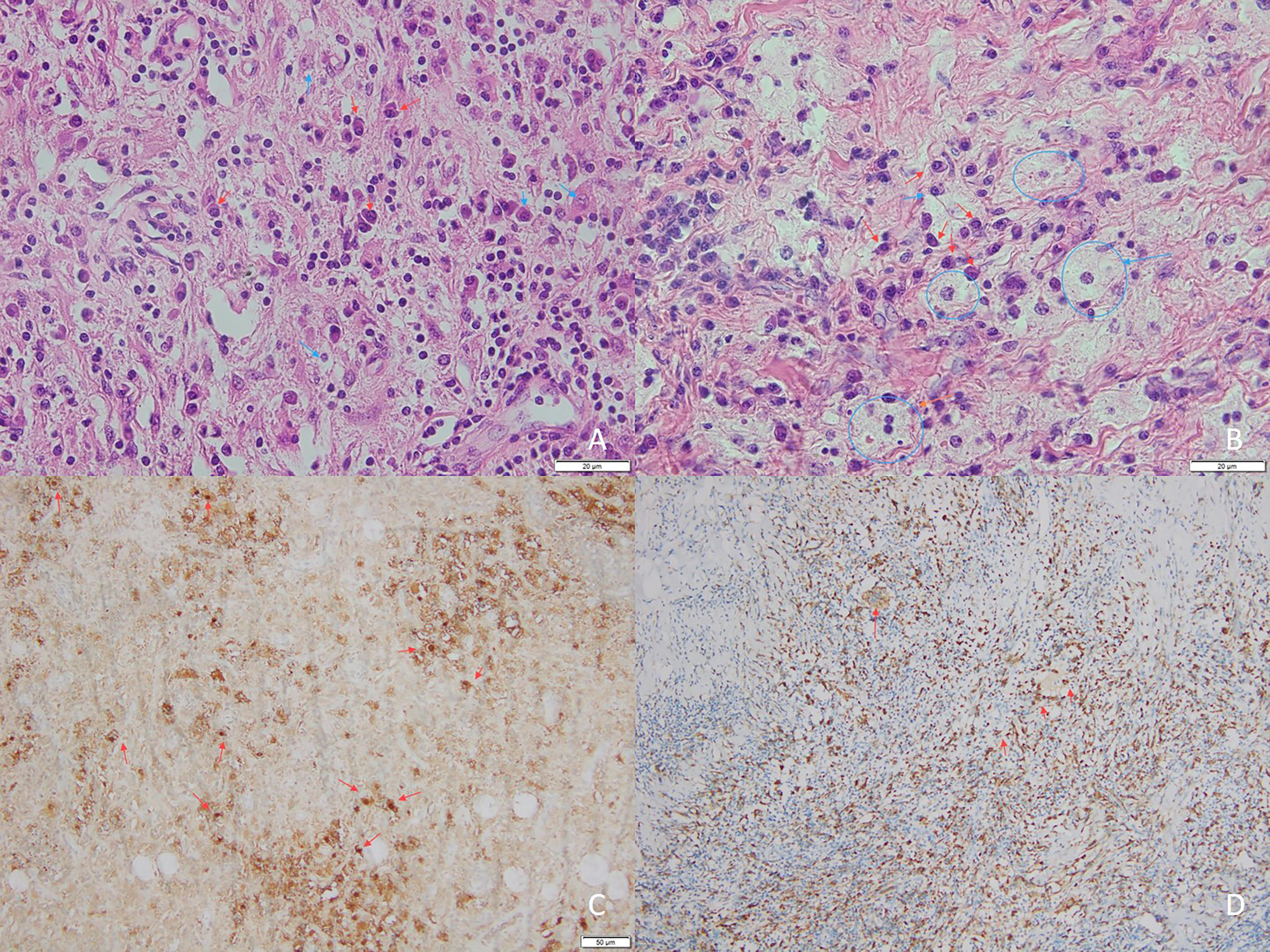
Histopathological examination of biopsy samples from all three cases showed the classical features of RDD. Hematoxylin and eosin (H&E) staining showed emperipolesis of plasma cells and lymphocytes, pale histiocytes, and collagen formation in the interstitium (Fig. 4A, B). The immunohistochemistry showed diffuse positivity for two classical RDD markers, Cluster of Differentiation 68 (CD68) and S-100, in the brain foci (Fig. 4C, D). The clustered S-100+ signals might be secreted S-100 in the intercellular matrix or damaged S-100-secreting cells. In addition, all three cases were negative for CD1a, Langerin, and Anaplastic Lymphoma Kinase (ALK) in their tumors.
WES findings
WES identified the mutation rs368284573 in the Phospholipase C Like 2 (PLCL2) gene across all three cases. The distribution and detection of this mutation within the target regions are illustrated in Figure 5. Figure 5A presents a proportional distribution map of the targeted exonic regions within the genome, demonstrating the relative concentration of target coverage. This visualization confirms efficient targeting for mutation detection by the densely covered on-target regions. The regions with a sequencing depth greater than 50×, as depicted in Figure 5B, constitute the predominant proportion. This high coverage depth indicates a higher confidence level in mutation detection within these segments, enhancing the reliability of our findings. The scatter plot of the segmented data from chromosome 3, where the mutation is shown in Figure 5C. Finally, the presence of the PLCL2 mutation was confirmed in Figure 5D through pyrophosphate sequencing, thereby confirming its occurrence within the targeted exonic region.
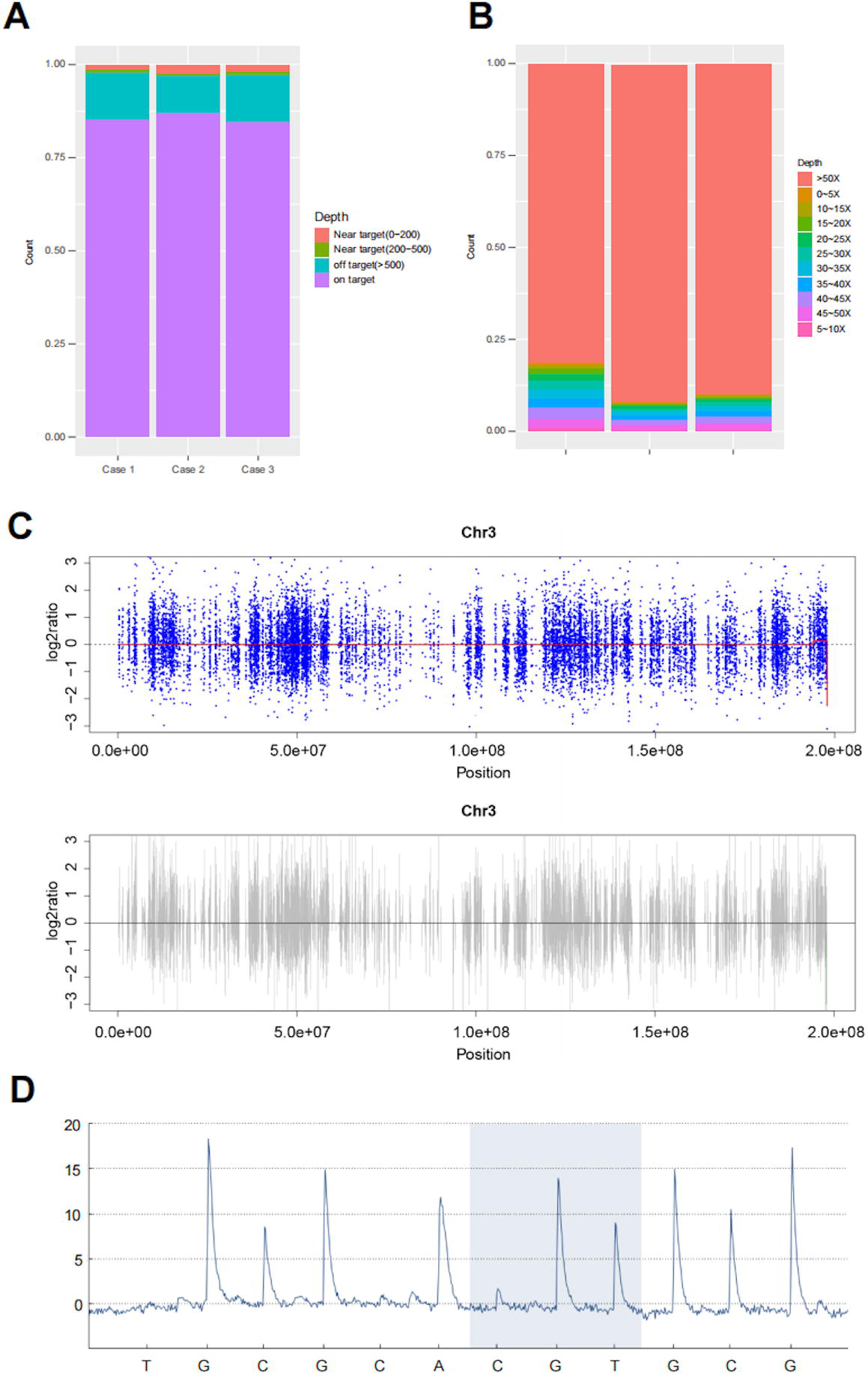
The detection of PLCL2 mutation sites, including
Functional analysis of PLCL2 mutation
PLCL2 mutation protects its protein from ubiquitination and promotes cell proliferation
To further explore the functional impact of the PLCL2 mutation, an overexpressed vector containing the mutant PLCL2 sequence was constructed. Figure 6A provides the sequencing results confirming the successful insertion of the mutated gene into the vector. Western blot analysis was then conducted to assess the protein stability of PLCL2 in 293T cells treated with the protein synthesis inhibitor cycloheximide (CHX). As shown in Figure 6B, the mutant protein exhibited enhanced stability under CHX treatment, suggesting a decrease in its degradation rate. Subsequently, we sought to ascertain whether the augmented stability of PLCL2 proteins resulting from the mutation can be attributed to attenuated ubiquitin–proteasome-mediated degradation. The Western blot results in Figure 6C demonstrated higher levels of PLCL2 protein and lower levels of ubiquitinated PLCL2 protein in cells harboring mutant PLCL2 constructs, when compared with the wild-type control group. To assess the impact of this mutation on cell proliferation, the cell counting kit-8 (CCK-8) assay was performed after silencing the mutant PLCL2 using RNA interference. Figure 6D illustrates that the PLCL2 mutation facilitated the proliferation of 293T cells, which was reversed upon the knockdown of the mutant PLCL2. These results reveal that the PLCL2 gene mutation protects the PLCL2 protein from ubiquitination, thereby increasing its protein levels and exerting a promoting impact on cell proliferation.
PLCL2 promotes the β-catenin signaling and cell proliferation by inhibiting the activity of GSK-3β
Given the essential role of the GSK-3β/β-catenin pathway in promoting cellular proliferation, 15 we examined the protein levels of key molecules in this pathway following the PLCL2 mutation. Subsequent to the PLCL2 mutation, the level of β-catenin was increased, while the phosphorylation of GSK-3β was decreased (Fig. 7A). Additionally, this mutation elevated the expression of c-Myc (Fig. 7A), which is a direct downstream target of β-catenin and drives cell proliferation. 16 Conversely, the expression of E-cadherin, a molecule that binds to β-catenin and limits cellular motility, 17 was negatively affected by the PLCL2 mutation (Fig. 7A). Notably, these effects were reversed by siPLCL2 (Fig. 7A). Total GSK3β levels remain roughly the same across all groups, indicating that the PLCL2 mutation specifically affects GSK3β phosphorylation rather than overall expression. Next, the GSK inhibitor, thiadiazolidinone (TDZD-8) 18 was employed to investigate the role of GSK-3β in the downstream mechanisms of PLCL2. Western blot analysis revealed that TDZD-8 treatment effectively inhibited GSK-3β phosphorylation, promoted the β-catenin signaling pathway, and rescued the decreased levels of β-catenin and c-Myc induced by PLCL2 knockdown (Fig. 7C). In cell proliferation assays, TDZD-8 significantly alleviated the siPLCL2-mediated inhibition of cell proliferation capability (Fig. 7B). Collectively, these findings suggest that the elevated level of PLCL2 inhibits GSK-3β activity, thereby protecting β-catenin from degradation and activating proliferation-associated target genes.
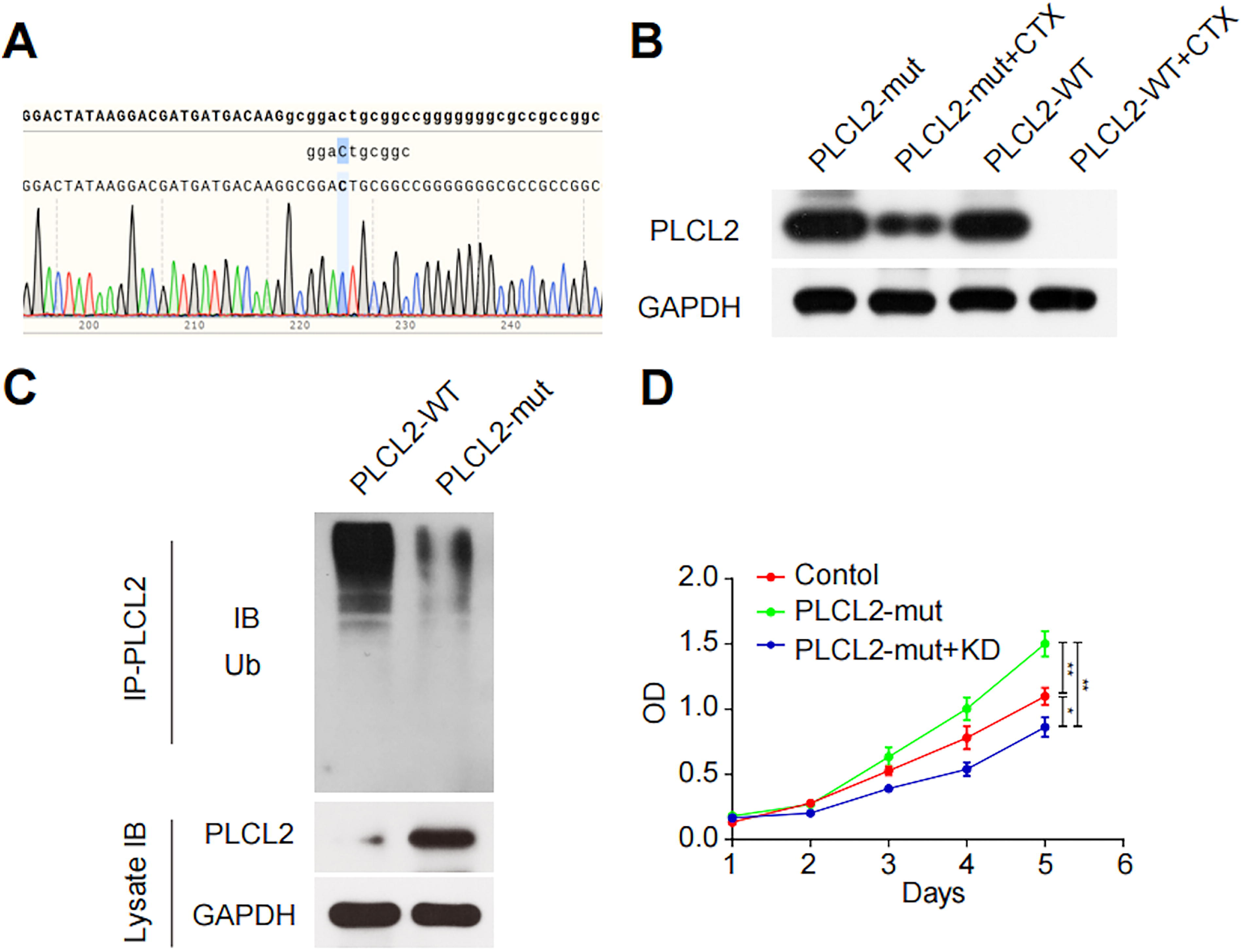
PLCL2 mutation protects its protein from ubiquitination and promotes cell proliferation.
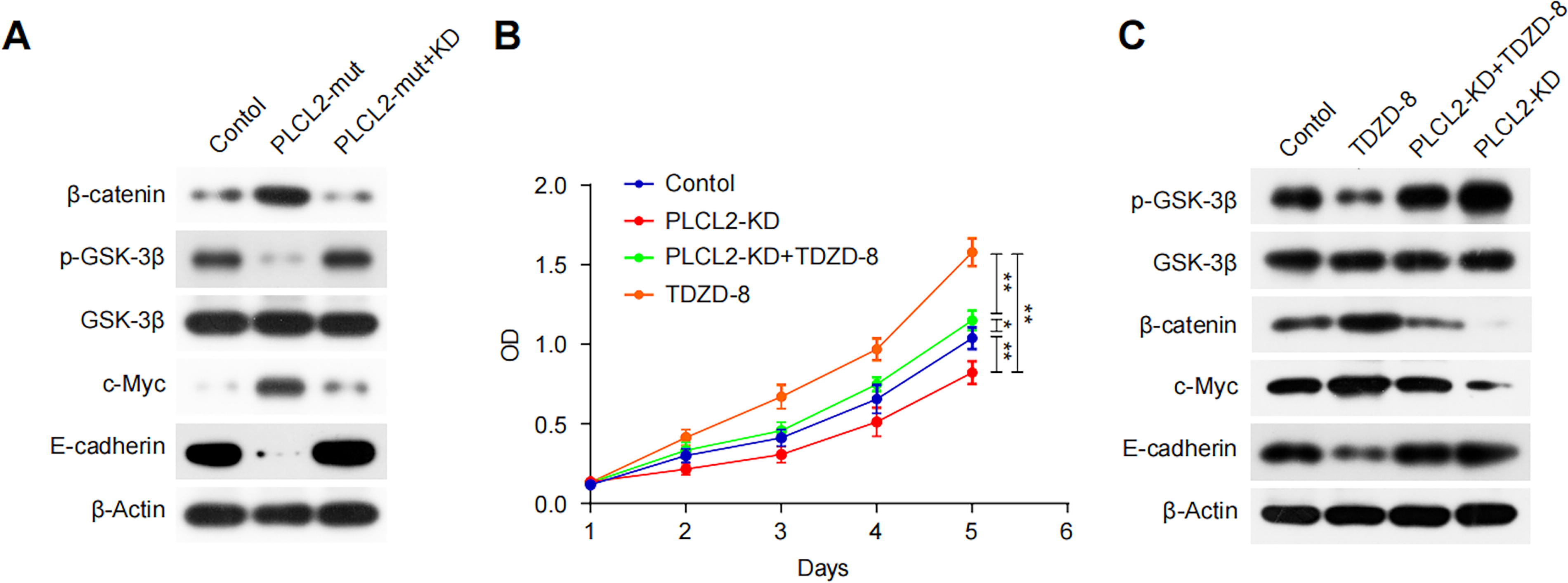
PLCL2 promotes the
Discussion
RDD, a rare form of non-Langerhans cell histiocytosis, was initially identified in 1965 by the French pathologist Pierre Paul Louis Lucien Destombes. 1 The clinical phenotype of RDD is highly variable, occurring independently or in conjunction with autoimmune or malignant diseases, making the diagnosis at initial presentation challenging. 19 In a cohort of 64 RDD patients diagnosed at a tertiary referral center between 1994 and 2017, approximately one-third had recurrence following their initial treatment. 20 Therefore, elucidation of the molecular mechanisms underlying the development and progression of RDD is crucial for optimizing diagnostic and therapeutic strategies. This study investigated three cases of extranodal RDD using a comprehensive approach encompassing clinical, radiological, pathological, and genetic analyses to enhance the understanding of its diagnosis and pathogenesis. Through WES, we identified a shared mutation, rs368284573, in the PLCL2 gene across all cases. Subsequent functional analyses revealed that the PLCL2 mutation exerted an influence on protein stability and cell proliferation via modulation of the GSK3β/β-catenin signaling pathway.
The most common extranodal lesions in RDD involve the skin and subcutaneous tissues, followed by the respiratory tract and bones. 5 Lesions in the CNS, cardiovascular system, and retroperitoneum have been reported in rare cases of RDD.21–25 This study reports three cases of extranodal RDD, including one rare case of meningeal involvement and two cases of subcutaneous involvement at distinct sites. Extranodal RDD can mimic other neoplastic and inflammatory conditions on imaging. Meningeal RDD is often initially misdiagnosed as meningioma or lymphocyte-rich meningioma, owing to the characteristic dural tail sign, 26 which was also observed in our CNS case. The dural tail sign in RDD potentially arises due to the infiltration of histiocytic cells into the dural layers and surrounding tissues, which induces local inflammation and fibrosis. This inflammatory response may result in a contrast enhancement at the periphery of the lesion, similar to what is seen in meningiomas.27,28 MRI perfusion has been proposed to assist in distinguishing RDD from meningioma. 29 Additionally, RDD may also radiologically mimic chronic subdural hematoma. 30 While the other two cases we report presented as isolated subcutaneous masses, cutaneous RDD can also manifest as multiple lesions or scattered rashes. 20 The involvement of the chest wall in RDD is exceedingly rare and needs to be distinguished from other tumor-like lesions that could occur in the chest wall, such as hemangioma, metastatic tumors, and dermatofibrosarcoma protuberans. Although atypical, these radiological signs play an important role in identifying extranodal RDD. Recent studies have suggested that Positron Emission Tomography (PET)/CT may be an important tool in the diagnosis and evaluation of RDD. 31 Further investigation is needed to refine the differential diagnostic criteria for extranodal manifestations.
Histopathological examination remains crucial in distinguishing RDD from other histiocytic and inflammatory disorders. Typically, the morphological characteristics of RDD are defined by the presence of large histiocytes containing hypochromatic nuclei and abundant pale cytoplasm. 32 Emperipolesis, a pathological hallmark of RDD, refers to the presence of intracytoplasmic leukocytes or lymphocytes within histiocytic cells. 5 However, it is rarely observed in the context of inflammatory infiltrate and fibrosis in extranodal foci. A case report documented lymphocyte-phagocytic emperipolesis in the cerebrospinal fluid of a patient with CNS RDD, offering a potential diagnostic insight for CNS RDD. 33 While uniformly round nuclei with smooth edges are commonly observed in these cells, occasional instances may exhibit nuclear irregularities and variations in cell shape. 34 The histiocytes in RDD display a phenotype characteristic of monocytes and macrophages, with positivity for CD68, S100 Calcium Binding Protein B (S100), Cyclin D1 (cyclin D1), POU Class 2 Homeobox 2 (OCT2), and Spi-1 Proto-Oncogene (PU).1 and variable levels of factor XIIIa. 19 The majority of these cells express CD163; however, around 10% of cases show negativity for this marker. Additionally, the lack of CD1a, langerin, or ALK in RDD sections suggests the non-Langerhans feature.32,35 The pathological findings in our three cases are consistent with the aforementioned, reinforcing that the pathological report provides the gold standard for diagnosing RDD.
While RDD has traditionally been considered non-neoplastic, recent genetic studies have increasingly pointed to clonal origins in some cases, especially with mutations affecting pathways such as MAPK/ERK. 5 In previously reported cases, the most commonly identified mutations in RDD involve MAPK or KRAS 7 genes. Alterations in BRAF have also been observed in RDD, particularly in conjunction with other histiocytic neoplasms.36,37 Additional genetic alterations include IRF5 mutations, AKT copy number variations and point mutations, and SLC29A3 mutations.9,14,38 These genetic architectures provide a foundation for the application of targeted pharmaceuticals in the treatment of RDD.39,40 Using WES of RDD samples from three different sites, we identified for the first time the PLCL2 mutation in RDD, which results in PLCL2 proteins accumulation. Genetic polymorphisms in PLCL2 have previously been linked to ischemic stroke in patients with arterial atherosclerosis or metabolic syndrome.41,42 Additionally, the PLCL2 rs1372072 is reported to enhance the susceptibility of limited cutaneous systemic sclerosis. 43 However, PLCL2 has been poorly studied in histoproliferative entities. Aberrant activation of Phospholipase C Gamma 1 (PLC-γ1) and Phospholipase C Gamma 2 (PLC-γ2), which share structural homology with PLCL2, has been implicated in inflammation, immunity, and cancer. 44 Overexpression of PLC-γ1 is a predictor of metastatic progression in breast cancer. 45 While we confirmed that PLCL2 mutations promote cell proliferation, further mechanistic studies are required to fully explore its potential as a therapeutic target.
In our study, no mutations were detected in NRAS, 6 KRAS, 7 MAP2K1, 8 ARAF, 6 or SLC29A3 9 genes as previously reported in the literature. A study including 21 patients with RDD noted that the mutations were more common in the CNS or lymph nodes and were mutually exclusive, with either KRAS or MAPK mutations being present, but not both. 7 Therefore, we hypothesize that patients suffering from RDD from different anatomical sites or exhibiting distinct symptoms may carry distinct mutations, highlighting the necessity for the individualization of targeted therapies.
Our experimental results demonstrated that the accumulated PLCL2 proteins can inhibit GSK-3β activity. In the process of wound healing, glucocorticoid-activated Wnt signaling has been shown to activate PLCs on cell membranes and regulate GSK-3β activity via the protein kinase C (PKC) cascade, resulting in β-catenin accumulation. 46 Similarly, the effect of PLCL2 on GSK-3β activity may involve PKC activation, which requires further studies to validate. Furthermore, mutated PLCL2 exerts sufficient impact on GSK-3β to promote β-catenin-associated cell proliferation. Despite its widely recognized protumorigenic role, 47 β-catenin also plays a critical role in some benign diseases48,49 and is essential for physiological differentiation. 49 Thus, building on the identification of the PLCL2 mutation and its impact on GSK-3β/β-catenin signaling, there is significant potential for developing novel therapeutic approaches aimed at targeting these pathways in RDD. TDZD-8, a known GSK-3β inhibitor, has shown promise in rescuing β-catenin degradation and could be explored as a potential therapeutic agent in RDD cases harboring PLCL2 mutations. 50 Furthermore, the activation of PKC by mutated PLCL2 suggests that PKC inhibitors could offer an additional therapeutic angle to disrupt the abnormal signaling cascade. 51 These interventions may help manage the proliferative aspect of RDD, particularly in cases where the disease exhibits aggressive behavior. Under different pathological contexts, the effects of β-catenin signaling are subject to precise and complex mediation. 52 Notably, none of the cases included in our study experienced postoperative recurrence during follow-up. Therefore, further research is warranted to elucidate the prognostic value of PLCL2 mutations in RDD.
This study is subject to several limitations. First, the small sample size of this study may impact the generalizability of the findings. Second, although we employed the high-quality formalin-fixed, paraffin-embedded (FFPE) DNA extraction kit to ensure reliable WES results, a comparative analysis of different sample collection methods merits further investigation. Third, the in vivo functional analysis might not fully capture the mutation’s effects in RDD histiocytes, given that the cellular origin of RDD is still undetermined. Fourth, in vivo functional analysis might not fully capture the mutation’s effects in RDD histiocytes, given that the cellular origin of RDD is still undetermined. Future research should aim to explore the genetic pattern in RDD in larger cohorts with longer follow-up periods, preferably through multicenter studies. Moreover, it would be valuable to explore the broader spectrum of signaling pathways in which PLCL2 is involved in RDD, potentially guiding targeted therapeutic approaches as alternatives to resection.
The Bigger Picture
This study sheds light on the significant role of the PLCL2 mutation in RDD, revealing how it influences the GSK-3β/β-catenin signaling pathway and promotes cell proliferation. Understanding this molecular mechanism opens up opportunities for developing targeted therapies for RDD. Future research could focus on therapeutic strategies that modulate this pathway, such as using GSK-3β inhibitors or gene therapy. These findings not only enhance our understanding of RDD but also provide valuable insights into similar pathways in other diseases, paving the way for more personalized treatments and better clinical outcomes for patients with histiocytic disorders.
Conclusion
This study provides new insights into the pathogenesis of extranodal RDD by identifying a mutation in PLCL2 and demonstrating its impact on the GSK3β/β-catenin signaling pathway. Imaging and pathological findings underscore the diagnostic challenges associated with extranodal RDD in clinical practice. Moreover, the integration of molecular and protein analyses offers valuable mechanistic insights that could potentially inform targeted therapeutic strategies.
Materials and Methods
Patients
This study is a retrospective case series conducted at the Third People’s Hospital of Chengdu, Sichuan, China, from 2020 to 2023. It includes three patients diagnosed with extranodal RDD, confirmed through histopathological and immunohistochemical analysis. The study followed ethical principles outlined in the Declaration of Helsinki (as revised in 2013). Additionally, it received approval from the Medical Ethics Committee of The Third People’s Hospital of Chengdu (No. 2024-S-68) and obtained informed consent from all participating patients.
Clinical, radiological, and pathological data collection
Detailed clinical information, including demographic data, presenting symptoms, and treatment history, were systematically collected for each case. Radiological imaging, including CT and/or MRI, was reviewed to document the location and extent of lesions. Histopathological analysis was performed using H&E staining, along with immunohistochemical markers, to confirm the diagnosis of RDD.
Follow-up
All three extranodal RDD patients participated in standard postoperative follow-up evaluations, which were conducted until August 1, 2024. The outcome of each patient was recorded.
Sample preparation
DNA for WES was derived from FFPE samples. DNA extraction was performed using the QIAamp DNA FFPE Tissue Kit (Qiagen, Germany) in accordance with the manufacturer’s protocols. The quality and concentration of the extracted DNA were then assessed to confirm suitability for subsequent analysis.
WES and variant analysis
WES analysis was performed using the Illumina HiSeg250 platform (lllumina San Diego, CA, USA) with 150 bp paired-end reads. All reads were mapped against the hg38 reference genome using the Burrow–Wheeler aligner (BWA). The identification of variants was performed using a custom pipeline that utilizes the Genome Analysis Toolkit (GATK) Best Practices workflow.
Variants were filtered according to minor allele frequency ≥5% in the 1000 Genomes and Exome Aggregation Consortium databases. Variants showed in the controls and synonymous variants, assumed to be benign or likely benign, were also excluded. Frameshift, stop gain, and stop loss were considered damaging. The nonsynonymous variants were predicted to be damaging by annotation in the dbNSFP database. In detail, different prediction software [Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v2 (High-Diversity) (Polyphen2_HDIV), Polymorphism Phenotyping v2 (High-Variability) (Polyphen2_HVAR), Light Rail Transit (LRT), MutationTaster, MutationAssessor, Functional Analysis Through Hidden Markov Models (FATHMM), Protein Variation Effect Analyzer (PROVEAN), Meta-Support Vector Machine (MetaSVM), Meta-Logistic Regression (MetaLR), Mendelian Clinically Applicable Pathogenicity (M-CAP), Combined Annotation Dependent Depletion (CADD), fathmm-MKL] were used to assign scores for the synonymous variants. Considering that some software may lack annotations for certain mutations, for those with available scores, if half or more of the software predict damaging, the variant is considered deleterious. Then, the criteria of the American College of Medical Genetics and Genomics were used to determine the pathogenicity of variants, building on the automatic classification provided by VarSome (https://varsome.com/). Finally, variants for further verification were selected based on mutation category and literature data.
Cell culture
Human Embryonic Kidney (HEK)-293 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Corning Life Sciences) with 10% fetal bovine serum (Premium Select, R&D Systems) and 1% penicillin/streptomycin (ThermoFisher) at 37°C and 5% CO2. Cells were cultured at uniform density in 24-well plates (Corning) for all experiments.
Western blotting
Cells were harvested in lysis buffer containing Nonidet P-40 (Amresco, J619), protease inhibitor cocktail (Roche, P9599), and phenylmethylsulphonyl fluoride. The bicinchoninic acid Protein Assay Kit (Thermo, MA) was used to adjust the concentrations of protein samples. Blotted membranes were blocked in 5% skimmed milk in tris-buffered saline with tween, followed by incubation with corresponding primary and secondary antibodies.
Cell proliferation assay
(CCK-8 was used for cell proliferation detection. 293T cells were inoculated into a 96-well plate and incubated for one night. After 24 or 48 h, 10 μL of CCK-8 was supplemented to each well for 2 h. Finally, the final optical density was determined at 450 nm using a microplate reader.
Authors’ Contributions
Conceptualization: Y.L. and Q.T. Methodology: Y.L., W.Y., and Q.T. Validation: Y.C. and L.S. Formal analysis: Y.L., W.Y., J.L., and J.G. Resources: Y.L. Data curation: Y.L. Writing—original draft preparation: Y.L. and W.Y. Writing—review and editing: J.L. and Q.T. Supervision: J.L. and Y.L. Software: Z.F. and L.S. Investigation: Z.F. Project administration: Y.C. and J.G. All authors have read and agreed to the published version of the article.
Footnotes
Acknowledgments
The authors acknowledge the entire working team.
Author Disclosure Statement
All authors hereby disclose that they have no conflicting interests.
Funding Information
This research received no external funding.
Data Availability Statement
No new data were created.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work, the authors used (Grammarly, Grammarly Inc.) to improve English writing. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.