
Abstract
Background
Increased stress in healthcare workers (HCW) is a global crisis and few brief, scalable interventions exist to support HCW with stress, anxiety, and insomnia. Individual interventions impacting these multiple symptoms are needed.
Objective
To determine whether a noninvasive brainwave echoing acoustic neuromodulation intervention reduces perceived stress among HCW compared to waitlist control.
Methods
This was a parallel randomized controlled trial of acoustic neuromodulation vs waitlist control conducted at a large academic health system. Adult HCW with Perceived Stress Scale (PSS) ≥14 were included. Exclusions were factors that would compromise intervention delivery or current engagement with similar interventions. The intervention was a limited dose paradigm of Cereset Research™ Standard Operating Procedures, a noninvasive, closed-loop, brainwave echoing acoustic neuromodulation neurotechnology, composed of four, 36-minute acoustic neuromodulation sessions over 2 weeks. The primary outcome was change in PSS at 6-8 weeks. Secondary outcomes were anxiety (Generalized Anxiety Disorder-7) and insomnia (Insomnia Severity Index). Exploratory outcomes included validated measures of depression, fatigue, and subjective cognition. Group-level changes in outcomes were evaluated using linear mixed models.
Results
Of 144 participants (72 per group), 134 completed primary outcome assessment (67 per group). Participants had mean age 44.7 years [SD 11.6] and were 86.1% female (N = 124). Intention-to-treat analyses demonstrated mean PSS score reduction of 7.8 (SD 5.9) in the intervention group vs 1.2 (SD 4.1) among controls (difference between groups 6.6 points, 95% CI 4.9-8.2, P < 0.0001). Secondary and exploratory measures also demonstrated significant group-level improvements in intervention vs control, and there were no serious adverse events.
Conclusions
In HCW with elevated stress, acoustic neuromodulation resulted in clinically meaningful improvements in perceived stress, anxiety, and insomnia. The intervention is safe, scalable, and may merit adoption by health systems to complement organization-level approaches for enhancing HCW well-being. Trial Registration: ClinicalTrials.gov, NCT04682197.
Introduction
Well-being in healthcare workers (HCW) remains a critical challenge. The COVID-19 pandemic compounded underlying levels of stress, emotional distress, anxiety, compassion fatigue, and burnout even in settings where HCW were satisfied with organization-level support.1-3 Literature suggests a dramatic increase in reported stress, anxiety, insomnia, and depressive symptoms in HCW, with a need to develop both individualized and organizational strategies to support well-being and mental health.4,5 Physician burnout rates are over 50%, significantly higher than other US workers.6,7 Increased behavioral symptoms can lead to poor provider performance and medical errors. 8 Further, across a broad range of roles, HCW have more anxiety, depression, somatization, and insomnia than workers outside of healthcare. 9 With higher workloads and fewer resources, healthcare professionals across different disciplines are experiencing greater burnout and increased turnover.10,11 Thus, interventions should include all HCW, not just nurses and physicians.
Systemic approaches in health systems and hospitals have been used to address burnout and well-being. 7 While organization-level strategies are important, individual-level approaches offer potential benefit, and combinations of individual-level and organization-level strategies are necessary for the greatest impact. Previous strategies investigated to mitigate and manage stress in individual HCW include mindfulness,2,12,13 body-based therapies (yoga, massage), 14 cognitive behavioral therapy, 15 meditation/relaxation,16,17 biofeedback-assisted resilience training, and multi-modality integrative medicine interventions. 18 The majority involve conscious, cognitive effort of the individual, ongoing practice to achieve and maintain benefits, and may require significant time commitment. Even with some successes, challenges remain for implementation and system-wide adoption, especially time burden, access, and lack of durable results.13,19,20 Some strategies narrowly target specific symptoms, lacking broader impact across various behavioral symptom categories, and with no demonstrated benefit to autonomic nervous system. 21 There is a unique need in the well-being space to identify brief, durable, and safe ways to support HCW individually across domains of stress, sleep, anxiety, and autonomics.
Brainwave echoing acoustic neuromodulation offers an individually tailored approach to address current barriers to HCW well-being interventions. Cereset Research™ (CR, of Cereset, LLC, Scottsdale, AZ), is an FDA-exempt closed-loop, brainwave echoing, acoustic stimulation neurotechnology (hereafter acoustic neuromodulation) that monitors individual brain activity in real-time and makes dynamic therapeutic adjustments within milliseconds. This precision-guided technology and a more operator-dependent precursor technology (High-resolution, relational, resonance-based, electroencephalic mirroring, HIRREM®) demonstrated significant, clinically meaningful symptom score reductions for up to 6 months post-intervention for insomnia, anxiety, depression and post-traumatic stress in a variety of clinical samples including insomnia, PTSD, athletic TBI, POTS, and menopause.22,23 Recent work demonstrated similar efficacy of scalable standardized operating procedures for this technology. 24 While the mechanisms are not definitively known, they may include resonance between the acoustic stimulation and brain oscillations. 23 Prior work also demonstrated sustained improvements in autonomic nervous system function to 4 months post-intervention 22 and improved network connectivity on fMRI. 25 This form of acoustic neuromodulation requires no conscious, cognitive activity on the part of the recipient, making it especially attractive to reduce stress in HCW.
Objective
Acoustic neuromodulation may meet the need for individualized, brief, non-effort-dependent, durable interventions to decrease HCW stress. The objective of this study was to evaluate a 2-week, 4-session standardized acoustic neuromodulation intervention for its impact on HCW experiencing self-reported moderate to severe perceived stress. This trial was designed to test for a moderate, clinically meaningful improvement in perceived stress (Perceived Stress Scale, primary outcome) at 6-8 weeks from baseline among HCW randomized to acoustic neuromodulation vs waitlist control.
Methods
Study Design, Ethics, Clinical Trials Registration
This trial used a parallel group, randomized controlled design with participants randomized 1:1 to the Cereset Research acoustic neuromodulation intervention vs waitlist control conducted at a single large healthcare system in the Southeastern United States. The study was approved by the local IRB and registered at clinicaltrials.gov (NCT04682197). All participants completed full informed consent prior to participation. This report focuses primarily on pre-specified primary and secondary outcomes.
Participants
Adults with moderate to high perceived stress (score on 10-item Perceived Stress Scale ≥14) 23 who were paid workers in any healthcare setting role were recruited via advertisements within the health system, information sessions, and word of mouth. Any employee at the medical center, including administrative, research, and non-clinical positions, were eligible. Exclusions were: factors precluding intervention delivery (unable to sit in a chair for up to an hour, weight over 400 pounds, insufficient hearing to receive intervention), previous use of the intervention, current participation in another research study with an active intervention (by medication or similar modality), current therapy with similar interventions [electroconvulsive therapy (ECT), transcranial magnetic stimulation (TMS), transcranial direct current stimulation (TDCS), alpha stimulation, eye movement desensitization and reprocessing (EMDR), brain spotting, neurofeedback, biofeedback, or deep brain stimulation (DBS)], current suicidality, seizure disorder, or current and significant symptoms of long-COVID. Importantly, there were no medication or substance use exclusions as in previous studies evaluating the technology.
Patient and Public Involvement
Nursing leadership was consulted on study design and practical aspects of implementing the intervention in HCW. For recruitment, marketing materials were shared, and in-services were done for nursing and institutional leaders. The public was not involved in the design, conduct, reporting, or dissemination plans of this research.
Intervention
Individuals allocated to the Intervention group received four, 36-minute brainwave echoing acoustic neuromodulation sessions, consisting of four individual protocols, each typically lasting from 8-12 min. Protocols were completed in pairs to limit interruptions and sensor placement changes. The software automatically switched from one pair to the other between protocols reducing the number of sensor placement changes needed during a session. Sensors monitored dominant brainwave frequencies at that location and translated them to audible tones in real time. Only one pair of sensors were actively echoing feedback. Protocols were administered with four sensors (two, paired locations) placed on the scalp at a time. Sensors were placed symmetrically at predetermined scalp locations, targeting the bilateral hemispheres according to the 10-20 International System at FP1/FP2 and T3/T4, F3/F4 and P3/P4, and C3/C4 and O1/O2. 26
The four sessions were to be delivered over a 2-week period, with the first two sessions occurring within the first 5 days, and all sessions completed within 10 business days. Total listening time was 144 min. Sessions occurred with eyes closed; participants were instructed to relax while sitting or reclining in a chair (Human Touch PC-6). Additional details regarding this standardized acoustic neuromodulation intervention has been previously described in detail. 24
Individuals assigned to waitlist control were offered intervention following primary outcome collection (6-8 weeks after enrollment). Participants in both groups continued current medical/psychological care.
Data Collection and Outcomes
Following electronic eligibility screening, informed consent and baseline data collection occurred in a research office, in a separate area of the research office from where intervention delivery occurred. At baseline (Visit 1, V1), demographics, health and work history, and baseline outcome measures were collected. Follow-up outcomes were collected 2 weeks after baseline (immediate post-intervention Visit 2, V2 visit) and the main post-intervention outcomes were collected 6-8 weeks after the baseline visit (Visit 3, V3, 4-6 week post-intervention visit). Baseline and follow-up data were collected by direct participant self-report using REDCap 27 surveys, usually completed in person.
Primary Outcome Measure
The Perceived Stress Scale (PSS) is a ten-item self-report measure with items rated from 0-4 and scores <14 indicating low, 14-26 moderate, and 27-40 high perceived stress. 28 A clinically relevant difference is 4 points. 29
Secondary Outcome Measures
Insomnia was measured using the Insomnia Severity Index (ISI), a 7-item scale with items scored 0-4 and these total score interpretations: 0-7 no insomnia, 8-14 subthreshold insomnia, 15-21 moderate insomnia, 22-28 severe insomnia.30,31 Clinically meaningful change is a 6 point reduction. 32 The Generalized Anxiety Disorder-7 (GAD-7), is a seven-item validated anxiety measure with total scores from 0 to 21 and the following interpretations: 0-4 minimal anxiety, 5-9 mild anxiety, 10-14 moderate anxiety, and 15-21 severe anxiety. 33 Clinically meaningful change is a drop of at least 4 points. 34
Exploratory Outcome Measures
Depression was measured using the Center for Epidemiologic Studies Depression Scale (CES-D) a 20-item depression screener 35 ; scores range from 0-60, with scores ≥16 indicating clinically relevant symptoms. 36 Perceived cognitive dysfunction was measured using the Multiple Ability Self-Report Questionnaire (MASQ), 37 a 38 item self-report questionnaire assessing language, visual/perceptual ability, verbal memory, visual memory, and attention. 38 Fatigue was measured using the Fatigue Severity Scale (FSS), a nine-item instrument assessing how fatigue interferes with daily activities. With total scores ranging from 9 to 63, and higher scores indicating greater severity. 39
Sample Size
A total of 144 participants were enrolled to achieve the pre-specified target of 138 study completers, defined as those who completed V2 (V2) data collection in both study arms. The trial was powered to detect a between-group difference of 3 points in change scores on the Perceived Stress Scale (PSS) from baseline to 6-8 weeks (V3; primary outcome). Sample size calculations were based on preliminary data from the study team’s prior work, which estimated a standard deviation of 5.62 for the change in PSS scores from V1 to V3. Assuming 80% power and a two-sided alpha level of 0.05, the minimum required sample size was 56 participants per arm. The final enrollment exceeded this threshold to ensure adequate power for secondary analyses and to account for potential attrition.
Randomization and Blinding
After completing baseline assessments, participants were randomly allocated 1:1 to intervention or waitlist control using a blocked randomization scheme with a block size of four uploaded in the data collection system by the team member who created it (who had no role in enrollment or assigning to intervention). The randomization scheme was concealed from other team members within the REDCap system until baseline data collection was complete and the enrolling coordinator then pressed the randomization button for intervention allocation. As this was a waitlist control study design, participants were not blinded to group allocation. The team study coordinator who initiated participant outcome surveys (for self-completion by participants, usually on a computer in the research office) was also responsible for intervention scheduling coordination, and was thus not blinded to group allocation.
Statistical Analyses
Descriptive statistics, plots, and histograms were used to examine distributions. Group-level differences in estimated mean change scores from baseline to 6-8 weeks were evaluated using linear mixed-effects models implemented via SAS PROC MIXED, with adjustment for baseline values of the outcome variable. This approach accounts for within-subject correlation and allows for the inclusion of participants with incomplete follow-up data, thereby enhancing the robustness of the longitudinal analysis. Goodness of fit was assessed using residual analysis; the residuals were randomly distributed, with no indications of heteroscedasticity or non-linear relationships. Intention-to-treat analyses were conducted. To control the family-wise error rate (FWER) across multiple hypothesis tests, we applied the Holm-Bonferroni correction. 40 This sequential method adjusts P-values by ordering them from smallest to largest and comparing each to a progressively less stringent threshold. Hypotheses were rejected sequentially until a P-value exceeded its threshold, ensuring strong control of Type I error while maintaining statistical power. There were seven outcomes assessed, and all were below the threshold P-value for significance. The proportion of individuals who achieved clinically meaningful improvements in the primary and secondary outcomes were calculated for each group and compared using Fisher’s exact test. Demographics of individuals who did not complete the intervention/study follow-up, and those who did not complete the intervention per protocol were compared to completers using Fisher’s exact and two sample t-tests. A P-value of <0.05 was considered statistically significant. All analyses were performed using SAS, version 9.4 (Cary, NC, USA).
Safety Assessment
In both groups, the study coordinator ascertained adverse events and any minor side effects via an open-ended question (“Have you noticed any positive and/or negative changes that we haven’t discussed or that I’ve asked today that you’d like me to write down?”) at data collection visits. Responses were written in the study file and categorized in the study database as one of four categories of pre-specified expected adverse effects (sleep, emotional changes/irritability, head fullness, or fatigue) vs other effects, with specific symptom specified. Other adverse events were identified via notification of emergency or hospital visits to the study team by the electronic health record. These were followed to resolution and discussed with the primary investigator, as were any unexpected adverse events.
Results
Participant Recruitment and Follow-Up
A total of 257 individuals were assessed for eligibility, 168 met PSS criteria, and 144 adults enrolled between December 2021 and June 2023 (Figure 1). Nearly one-third of those not enrolled had incomplete screening forms (n = 35/113, 31%), and 36 (32%) were ineligible. Nearly half of these (n = 17) were ineligible due to low PSS scores, and eight were excluded due to active suicidal ideation in the past month. Consort Diagram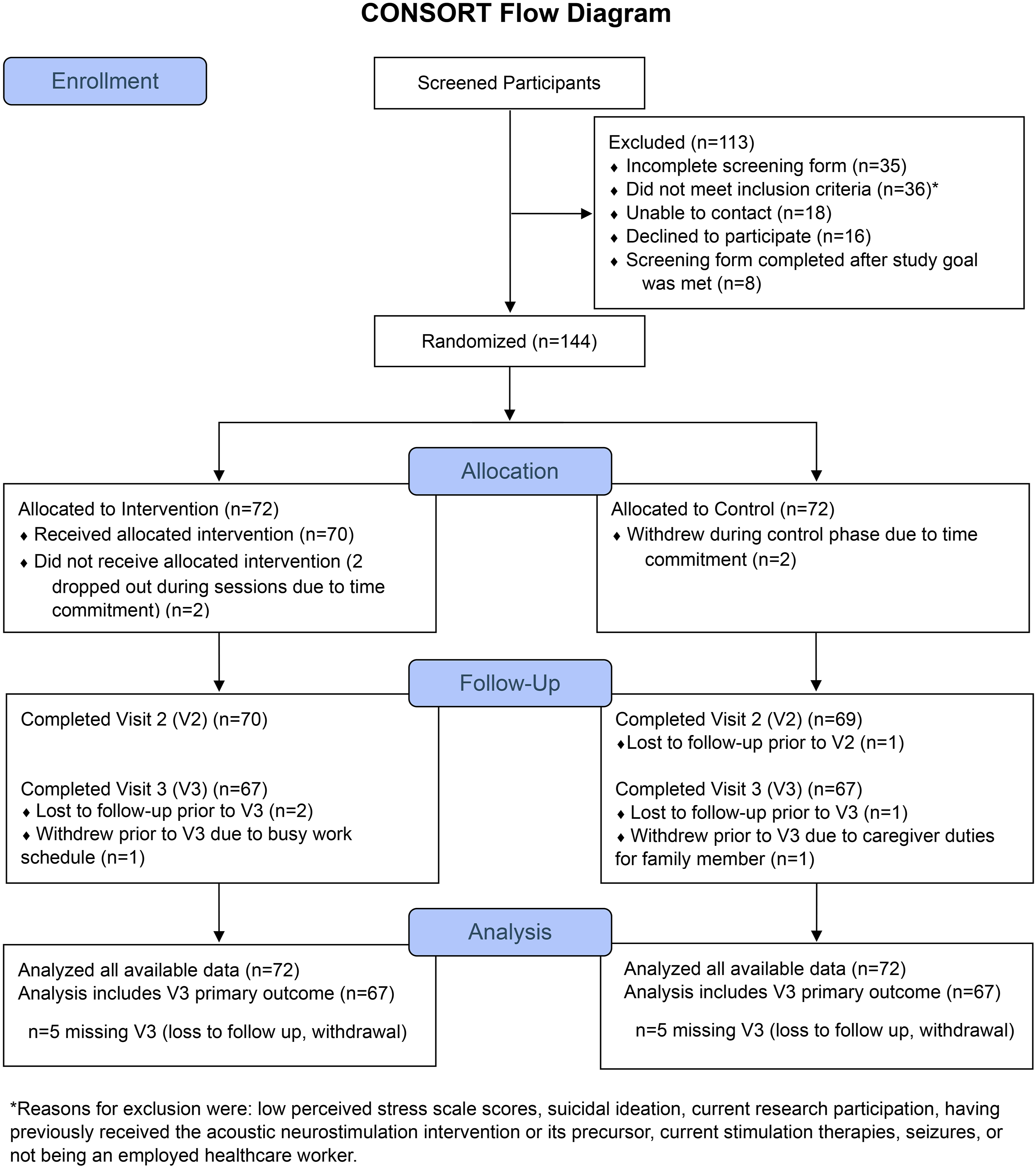
Participant Baseline Characteristics
Demographic and Clinical Characteristics of Participants

Notes. SD: standard deviation; IQR = interquartile range;
aShift schedule categories are not mutually exclusive;
bIncludes Thai Chi, mindfulness, and music for one individual each.
Outcome Collection and Intervention Delivery
Data collection occurred from December 2021 to August 2023. Three participants in each group withdrew due to time commitment or competing demands, and two in each group were lost to follow-up, leaving 67 individuals with full outcome data available at the primary outcome assessment (Figure 1). Outcome assessments occurred a mean 16.8 (SD 6.0) days after enrollment for V2, and 53.7 (SD 10.2) days for the V3 main outcome. In the intervention group, seven participants did not complete the four intervention sessions according to study protocol.
Primary and Secondary Outcomes: Group Level
Group-Level Outcomes: Primary, Secondary and Exploratory

Notes. SD: standard deviation; LSMean: least squares mean; SE: standard error; V2: Visit 2; V3: Visit 3; PSS: Perceived Stress Scale; ISI: Insomnia Severity Index; GAD-7: Generalized Anxiety Disorder-7; CES-D: Center for Epidemiologic Studies Depression Scale; MASQ: Multiple Ability Self-Report Questionnaire; FSS: Fatigue Severity Scale.
aN = [baseline, V2, V3, change from baseline to V3]; V2: immediately post-intervention (2 weeks after baseline); V3: 4-6 weeks post-intervention (6-8 weeks after baseline); analyses all by original assigned groups.

Group-Level Change in Perceived Stress, Anxiety, and Insomnia From Baseline to V3. PSS: Perceived Stress Scale; ISI: Insomnia Severity Index; GAD-7: Generalized Anxiety Disorder-7; V1: Visit 1; V2: Visit 2; V3: Visit 3; N = 72, 70, 67 in Intervention Group and 72, 69, 67 for Waitlist Control at V1, V2, and V3 Respectively
Group-Level Changes in Exploratory Outcomes
Table 2 also demonstrates changes in depression, perceived cognition, and fatigue scores. The intervention group had statistically significant greater improvement at V2 compared to waitlist controls. Using the Holm-Bonferroni correction, all outcomes are statistically significant; our largest P-value was P < 0.001 for the MASQ score, which was well below the threshold of 0.05 for the test of significance for this measure.
Individual Level Changes in Primary, Secondary Outcomes
Figure 3 shows the distribution of participants across symptom severity categories for the primary and secondary outcomes at baseline and follow-up. In the intervention group, the severity distribution transitioned toward milder scores across all three measures, with the majority of intervention participants scoring in the mildest category by V3, while score severity distributions appeared stable in the control group. Regarding individual-level clinically meaningful improvement, a ≥4 point improvement in PSS occurred in 72% of intervention participants, compared with 27% of controls (P < 0.0001), Fisher’s exact test. Similarly, a significantly higher proportion of intervention participants had meaningful anxiety and insomnia changes by V3 compared to control. Individual-Level Changes in Severity of Perceived Stress (A), Insomnia (B), and Anxiety (C) During Follow-Up. PSS: Perceived Stress Scale; ISI: Insomnia Severity Index; GAD-7: Generalized Anxiety Disorder-7; V1: Visit 1; V2: Visit 2; V3: Visit 3
Sensitivity Analyses Including per-Protocol Sample
Overall, six individuals did not complete the V2 follow-up (three per group; non-completers). The three non-completers in the intervention group did not complete all four intervention sessions. Non-completers and individuals with missing outcome data (N = 5 per group) were not significantly different in terms of race, ethnicity, shift schedule, baseline perceived stress, anxiety or insomnia scores from those who completed the initial 2-week intervention/control period or had complete outcome data (Supplemental Tables 1-2). Also, these characteristics were similar to those who completed the intervention per protocol among the seven intervention group participants who completed four intervention sessions but deviated from study protocol in timing between intervention sessions.
Safety
There were no serious adverse events or any unexpected intervention-related adverse events. Two participants had planned surgeries during the study, and four individuals had unrelated medical issues identified (back pain, ovarian cyst pain, celiac symptoms, migraine). Other minor, temporary side effects were reported to the study coordinator at the V2 or V3 outcome visit in response to the open-ended question detailed in Section 2.9. At these timepoints, 21 individuals total in the intervention group (29.2%; 15 individuals at only V2, three individuals at only V3 and three individuals at both timepoints) and four individuals in the control group (5.6%; one individual at V2 only, two individuals at V3 only, and one at V2 and V3) reported an adverse event. All of the minor side effects were expected (the four pre-specified symptoms and headache, which had not been prespecified but was not deemed unexpected); one unexpected other symptom was reported—increase in hot flashes, in the control arm. Minor side effects included sleep changes which were reported by 18% of the intervention group (N = 13) and 2.8% of controls (N = 2); and headache, reported by 11% of intervention participants (N = 8) and 1.4% of controls (N = 1). Less often, emotional changes/irritability (N = 3 in intervention, 4.2% and 1 control, 1.4%), head fullness (N = 3 in intervention, 4.2%, 0 controls), and fatigue (N = 2 intervention, 2.8% and 1 control, 1.4%) were reported. These symptom reports were consistent with known, temporary physical sensations, and minor side effects of the intervention.
Discussion
In this randomized trial of a scalable, less operator-dependent limited-dose brainwave echoing acoustic neuromodulation, HCW reporting elevated stress levels completed four intervention sessions over 2 weeks. Compared to waitlist control, participants in the intervention group showed clinically meaningful and statistically significant reductions in perceived stress, insomnia, and anxiety at 6-8 week follow-up. The current clinical findings are consistent with a previous randomized, controlled trial for insomnia using legacy technology, 22 as well as a smaller trial evaluating a prior, more scalable version of the intervention for insomnia.22,24 Compared to the legacy technology, HIRREM, the current technology uses updated hardware, and software, and faster computers and processing speed. Prior studies using HIRREM typically required eight to twenty sessions, with listening times of 60-90 minutes per session. A previous study with Cereset Research Standard Operating Procedures included ten sessions, with a total listening time of 536 min. This study expands on prior work by evaluating an even briefer standardized approach, with only four intervention sessions, decreasing the participant burden with shorter (only 144 total minutes listening time over the four sessions), and fewer sessions. The use of a standardized sequence of scalp locations during each session reduced intervention administration time. Newer computers enabled more rapid brain echoing which supported enhanced brain response requiring fewer sessions. This is also the first study evaluating this acoustic neuromodulation technology that did not have exclusions for various classes of medications, alcohol, or recreational drugs. The results suggest a clinically meaningful benefit for those taking psychotropic medications. This study is an important step forward that offers greater potential for scalability and generalizability. The findings indicating improvement in the secondary outcome of insomnia further adds to a growing body of literature suggesting a benefit of acoustic stimulation to treat insomnia. 41
We recognize that work on system-level interventions is critical to impacting well-being in HCW; such interventions are needed to focus in multiple identified target areas (culture of well-being, efficiency of practice, personal resilience).3,7 Given that system-level interventions take longer to implement and realize cultural shifts, 42 efficacious individual interventions are important and can provide more immediate relief. Numerous non-pharmacological strategies have been tested for HCW with symptoms of stress, anxiety, and insomnia. Some common approaches include education, mindfulness, CBT, massage, and yoga.13,14,43 A key difference between these other interventions and the acoustic neuromodulation intervention tested in this study is time commitment.16,44 Additionally, the current study evaluated closed-loop, recipient unique acoustic neuromodulation technique engaging the brain to harmonize and balance itself. Many studies have high dropout rates due to large time commitments 2 or involve active, conscious participation. The current intervention does not require cognitive engagement45-47; rather, participants are encouraged to relax or fall asleep.
Several key differences between this intervention and these other approaches include the use of a closed-loop paradigm, a relatively brief intervention period, lower dropout rates, positive changes across symptom clusters vs one area, 2 and clinically meaningful change in the PSS, ISI, and GAD-7 outcome measure. This study also demonstrated robust changes across individual level severity categories of scores. We recognize that there are limited data on closed-loop, non-pharmacological modalities for a true comparison to this study.
Next Steps
Because the limited dose intervention was well tolerated and feasible in a cohort of busy HCW, and showed short term benefits, with few exclusions, future studies should be conducted with extended follow up to evaluate long-term benefits of acoustic neuromodulation. Since HCW have continuous exposure to stressors, it will also be important to explore if periodic additional sessions can extend benefits. As healthcare professionals continue to struggle with well-being and self-care, drastic efforts are needed to manage stress. Scaling and implementing scientifically validated strategies will help prolong career trajectory and prevent burnout and care-providers leaving the medical field while also potentially improving business outcomes. Health system relevant outcomes include personal factors such as absenteeism and healthcare utilization, whereas system-wide priority metrics could be asked in annual surveys with a focus on culture or safety results. While health systems must also continue to identify and implement strategies focused on systemic issues that contribute to ongoing HCW stress, these results suggest that acoustic neuromodulation might offer an important individual level intervention to help mitigate the effects of stress in the healthcare setting. The authors hypothesize that there could be synergistic benefits to multiple realms of health outcomes using this intervention along with organizational strategies to promote well-being and engagement. 7
Limitations
Study limitations potentially impacting generalizability include scheduling challenges with HCW, as participants regularly rescheduled their sessions. Because sessions were offered during weekdays from roughly 7 AM to 6 PM, shift workers may not be adequately represented. Despite the limited dose, some HCW could not participate due to the time commitment. Recruiting primarily from the health system could have introduced selection bias. Participants were mostly females and nurses. Future studies should include a broader range of HCW roles for increased generalizability.48,49 People who enrolled might have been more open to modalities to help manage stress than general HCW. Medications, alcohol, and recreational drugs were not excluded, so potential interactions with response or durability may not have been appreciated. The study was relatively short, with outcome measures only at 6 weeks post-intervention. This paper only reports on self-report symptoms. Lack of access to health record data is a limitation, and this precluded analyses regarding any potential effects related to clinical diagnoses or comorbidities. There was no objective sleep measure. Papers on objective autonomic changes and exploratory measures are forthcoming. We acknowledge that the waitlist control design and lack of blinding are significant limitations of the trial; these design decisions were made primarily due to resource considerations, as there were not sufficient resources to conduct a sham arm or add an additional study coordinator to serve as a blinded assessor. We agree there is a potential bias posed by the lack of blinding in this trial, though we have previously demonstrated benefit in sham-controlled trials of the precursor (HIRREM) technology.22,24 Further, we recognize that the waitlist control design is a study limitation and may result in overestimation of intervention effect. 50 Future studies should incorporate sham control arms rather than waitlist arms and employ blinded outcome assessors to enhance rigor. We also acknowledge the mechanism of action for this technology is not well characterized and is an important area for future research. Finally, outcome measures collected do not directly address health system relevant business outcomes.
Conclusion
This randomized trial of a limited-dose acoustic neuromodulation intervention for HCW with elevated stress levels resulted in clinically meaningful improvements in perceived stress, anxiety, and insomnia. The intervention also demonstrated safety, and scalability. These results suggest potential for use by health systems, along with organization-level approaches, to reduce stress among employees, and enhance HCW well-being. Studies are needed to evaluate the effect of ongoing periodic intervention sessions, as well as health system relevant outcomes.
Supplemental Material
Supplemental Material - Randomized Controlled Trial of Acoustic Neuromodulation to Enhance Well-Being in Healthcare Workers
Supplemental Material for Randomized Controlled Trial of Acoustic Neuromodulation to Enhance Well-Being in Healthcare Workers by Catherine L. Tegeler, Heidi Munger Clary, Hossam A. Shaltout, Gregory B. Russell, Suzanne C. Danhauer, Charles H. Tegeler in Global Advances in Integrative Medicine and Health.
Footnotes
Acknowledgements
In memoriam of Lee Gerdes, of Brain State Technologies, LLC and Cereset, LLC, Scottsdale, AZ. The authors thank Lee Gerdes for his vision, innovation, and long-standing collaboration, as well as assistance developing study design, protocols, and interpretation of data. The authors acknowledge Lindsay J. Howard, Faiza Asif-Fraz, Lauren B. Tickle, and Kenzie L. Brown for their assistance with provision of the study intervention. Dawn C. Higgins assisted with recruitment, scheduling, data collection, organization, and preparation of data for analysis.
ORCID iDs
Ethical Considerations
This study received ethical approval from the Wake Forest University Health Sciences IRB (IRB# 00066997) on November 15, 2021. The current protocol is approved through July 6, 2026.
Consent to Participate
All participants provided written informed consent prior to participating.
Author Contributions
All authors have reviewed the manuscript and agreed to be accountable for all aspects of this work. Concept and design: Tegeler CL, Tegeler CH, Munger Clary, Shaltout. Acquisition, analysis, or interpretation of data: Tegeler CL, Munger Clary, Russell, Shaltout, Danhauer, Tegeler CH. Drafting of the manuscript: Tegeler CL, Munger Clary. Critical review of the manuscript for important intellectual content: Tegeler CL, Munger Clary, Russell, Shaltout, Danhauer, Tegeler CH. Statistical analysis: Russell, Munger Clary, Tegeler CL. Obtained funding: Tegeler CL, Tegeler CH. Administrative, technical, or material support: Tegeler CL, Munger Clary. Supervision: Tegeler CL.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by the research grant from The Susanne Marcus Collins Foundation, Inc., REDCap infrastructure for data collection and management was supported by NIH grant UL1TR00142.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.