
Abstract
Background
Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein–Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the “XMEN” title pathologies. The updated title, “X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect,” was proposed in 2020.
Objectives
To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.
Methods
Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.
Design
Immune re-evaluation done through flow cytometry and next-generation sequencing.
Results
Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.
Conclusion
We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.
Keywords
Introduction
Primary immunodeficiency (PID) disorders include over 250 heterogenous inborn errors of innate or adaptive immunity, which are predominantly single gene defects.1,2 The most common disorders of adaptive immunity involve the B-cell lineage.1,2 These antibody-deficiency disorders characteristically present with recurrent bacterial sinopulmonary infections, as well as increased risk of co-morbid autoimmunity, hypogammaglobulinemia, opportunistic infection, and malignancy.1,2 Improved genomic technologies over the past several decades have advanced our diagnostic capacity to identify previously unknown gene defects and, subsequently, more specific diagnoses and personalized management of a substantial immunodeficient patient population. 2
Antall et al first discovered a unique B-cell subset cluster of differentiation (CD) surface expression predominance in a small male cohort of selective anti-polysaccharide antibody deficiency (SPAD) with recurrent sinopulmonary and severe viral infections, autoimmune phenomena, and hypogammaglobulinemia (Table 1). 3 Patient 1 from that cohort was identified at age 5 with severe SPAD and he presented around 23 years later for re-evaluation of his recurrent infections and autoimmune complications, which gave an opportunity for reassessment using modern genomic techniques.
The above patient was identified with recurrent infections, autoimmune complications, absent pneumococcal titers (post-vaccination), and elevated CD5 B-cells (97%). 3 Clinical exome sequence analysis revealed a magnesium transporter 1 (MAGT1) gene hemizygous splice site mutation, in addition to reduced activator CD16 + CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression on flow cytometry. This MAGT1 mutation and reduced NKG2D expression of Patient 1, as well as CD5+ PID cohort clinical presentations in subsequent published cases in 2000 and 2005,3–6 are in the spectrum of the “novel human primary immunodeficiency” introduced by the National Institute of Allergy and Infectious Diseases (NIAID) in 2011. 7 The NIAID has recently proposed an update to their originally designated title, “X-linked immunodeficiency with magnesium defect, Epstein–Barr virus (EBV) infection, and neoplasia” (XMEN), 8 considering its variable phenotypic expression and advanced MAGT1 glycobiology understanding.9–11 Recent investigations of MAGT1 reveal its role beyond intracellular magnesium (Mg2+) transport, as a non-catalytic subunit of the oligosaccharyltransferase complex (OST) in asparagine N-linked glycosylation of immune regulatory glycoprotein receptors, suggesting a combined immune and glycosylation defect.9–11 We agree with the phenotypic flexibility and genotypic specification of “X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect.” 8 However, we propose that the CD5+ PID described by Antall et al should be included in this disease spectrum.2,3
CD5 B-cell predominant primary immunodeficiency from combined male patient cohorts.
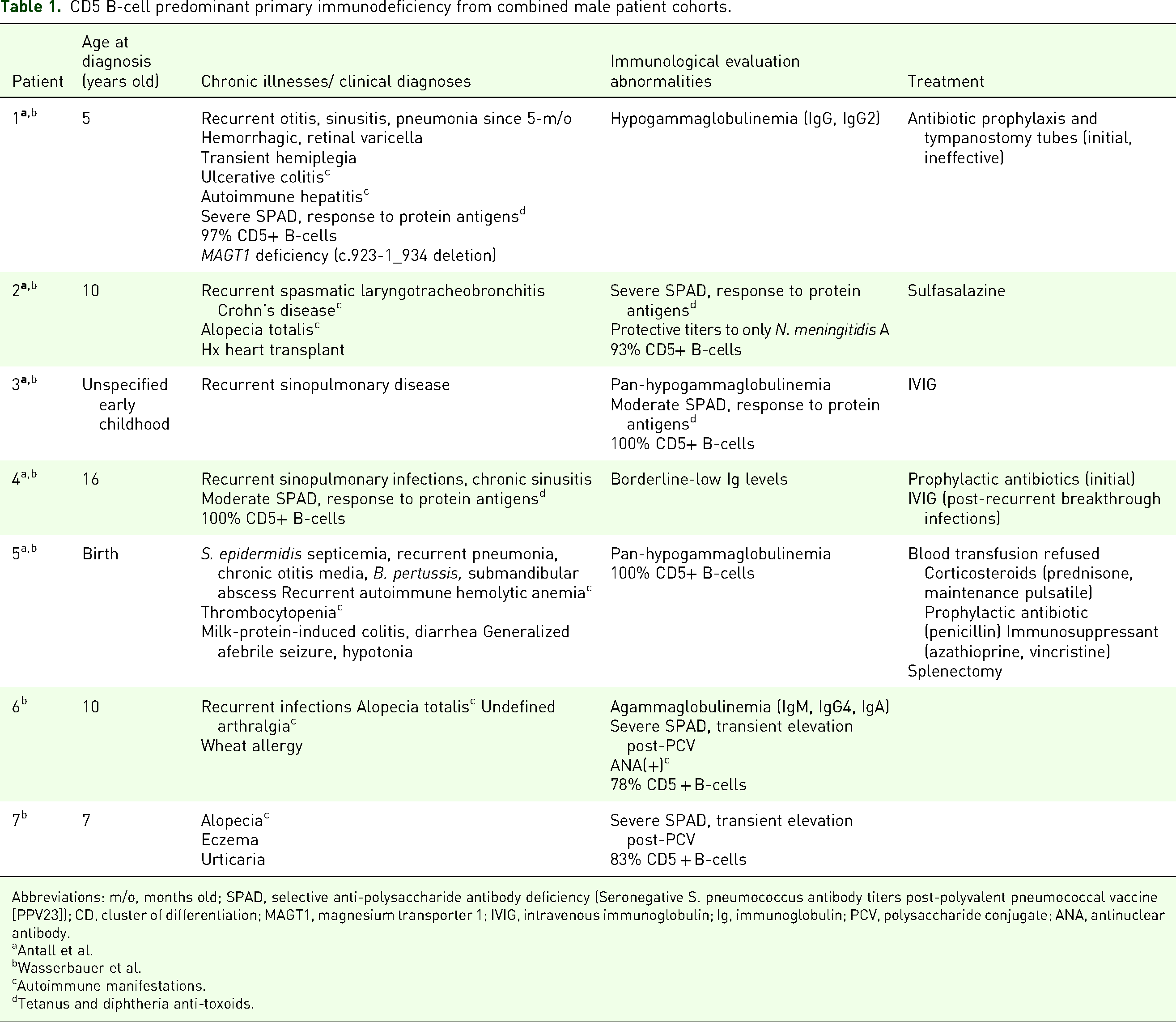
Abbreviations: m/o, months old; SPAD, selective anti-polysaccharide antibody deficiency (Seronegative S. pneumococcus antibody titers post-polyvalent pneumococcal vaccine [PPV23]); CD, cluster of differentiation; MAGT1, magnesium transporter 1; IVIG, intravenous immunoglobulin; Ig, immunoglobulin; PCV, polysaccharide conjugate; ANA, antinuclear antibody.
Antall et al.
Wasserbauer et al.
Autoimmune manifestations.
Tetanus and diphtheria anti-toxoids.
Methods
Patient History and Physical Examination
The study described is a single-patient study involving serological, flow cytometric, genetic evaluation over the course of several months. Verbal consent for the manuscript as well as written consent for flow cytometric studies were obtained from the patient. The majority of the studies that were performed were performed for diagnostic reasons and did not require IRB approval. The study that was experimental was related to the NKG2D receptor, and required IRB approval which was done through the NIH IRB (study number 06-I-0015) under the study title “Screening Protocol for Genetic Diseases of Lymphocyte Homeostasis and Programmed Cell Death.”
Patient 1 from the Antall et al cohort presented at 28 years old in August 1998 for further elevation of his peripheral blood CD5/CD19 B-cell predominance (97%, control 52.5 ± 17.3%) diagnosed at 5 years of age. 3 His initial immunological evaluation also revealed seronegativity of 12 Streptococcus pneumoniae subtypes (<0.3 µg/mL), Neisseria meningitidis A and C (<1/4), Haemophilus influenza B (<0.44 μg/mL), human parainfluenza viruses 1-3 (<1/8), influenza A and B (<1/40), poliovirus 1-3 (<1/8), and respiratory syncytial virus (<+2.0 SD). 3 Serum immunoproteins identified hypogammaglobinemia (Table 2). 3 Other immune laboratory assessment of blood leukocytes and mitogen- and antigen-induced lymphocyte proliferation panel was within normal limits. 3 Serologic evidence of human immunodeficiency virus, EBV, and cytomegalovirus were absent. 3 Post-vaccination antibody titers demonstrated sufficient (≥0.01 IU/mL) immunological response to tetanus and diphtheria anti-toxoids (Table 3). 3
Patient A immunoglobulin levels.

Patient A antibodies post-vaccination.

His past medical history was significant for the onset of recurrent sinusitis, otitis media, and pneumonia at 5 months of age. Tympanostomy tubes and prophylactic antibiotics failed to resolve his persistent childhood sinopulmonary infections. He was hospitalized for severe hemorrhagic varicella at 2 years of age, succeeded by acute hemiplegia, which self-resolved in 4 months and appeared non-pathologic on neuroimaging. He later developed autoimmune diseases, including ulcerative colitis in early adolescence and ongoing autoimmune hepatitis at 26 years of age. Unremarkable liver ultrasound did not necessitate medical management. Hypogammaglobinemia was persistent on routine labs. Family history revealed no known immunodeficiency. Review of systems and physical examination were unremarkable for acute concerns.
Immune Re-Evaluation
Flow cytometry. Immunophenotyping with monoclonal antibodies targeted against B-cell (CD19+) antigens was performed on aliquots of anti-coagulated (ethylenediaminetetraacetic acid) peripheral blood, as conducted in Antall et al 3 FACSDiva software (Becton, Dickson and Company© [BD] Biosciences, San Jose, CA) analyzed 100,000 cells processed through a FACSCantoTM II Cell Analyzer (BD Biosciences) within 12 h. Fluorochromes allophycocyanin (APC), BD HorizonTM violet laser 450/50 nm absorption/emission (V450) reagent, phycoerythrin (PE), and fluorescein isothiocyanate (FITC) were applied for maximal sensitivity. Distinct B-cell population percentages were quantified via dual-color fluorescence density plots by dividing a relative gated cell population in its dual-parameter quadrant of interest (left lower: double-negative; upper right: double-positive; upper left: single-positive (y-axis) CD antigen; lower right: single positive (x-axis) CD antigen) by the total.
PhiX174 (ΦX174) bacteriophage antigen immunization. Purified T-cell dependent neoantigen ΦX174 bacteriophage was intravenously administered (2 × 109 plaque-forming units [PFU]/kg bodyweight) as a primary immunization, according to methodology described by Och and Stiehm et al12,13 After circulating phage was no longer intravascularly detectable within 3-4 days post-administration, neutralizing antibody activity was weekly assayed via blood sampling. Secondary immunization was provided 6 weeks later. Antibody activity was calculated by standard rate of neutralization, or K value (Kv): K = 2.3*(d/t)*log(Po/Pt) (d, reciprocal of the serum dilution; t, reaction time, min; Po, phage titer at start, Pt, phage titer at time, t). 12 A neutralizing antibody specimen exposed to 2-mercaptoethanol A via gel filtration after each immunization distinguished between IgM (susceptible) and IgG predominance (resistant). 12
Next-generation sequencing. A primary immunodeficiency panel was ordered from a clinical diagnostic laboratory (Invitae Corporation©, San Francisco, CA) performing full-gene sequencing and deletion/duplication analysis using next-generation sequencing technology (NGS) to analyze genes associated with inherited immune disorders. Sequence analysis targeted covered non-coding variants and clinically significant coding exons, flanked by 10-20 adjacent intronic base pairs (bp) on either side. Deletion/duplication analysis determined the copy number at a single exon resolution at all targeted exons. Single nucleotide variants, insertions, and deletions and exon-level deletions and duplications less than 15 bp in length offered negligibly higher analytical sensitivity and specificity (>99%) than broader insertions and deletions within full exon length. The assay did not verify complete resolution and detailed analysis of specific structural rearrangements, such as inversions, gene conversion events, or translocations, nor variants embedded in complex genomic architecture, for example, short tandem repeats or segmental duplications. mosaicism, phasing, or mapping ambiguity.
Flow cytometric histogram with medical fluorophore 1 quantification. Medical fluorophore 1 (MF1) quantification of CD16 + CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein (Figure 3) was performed. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque PLUS (GE Healthcare) gradient density centrifugation. Red blood cells were removed by ACK lysis (Quality Biological). PBMCs were incubated with human Fc binding inhibitor (eBiosciences) in phosphate-buffered saline (PBS) at 25 °C for 20 min. Cells were then stained in ice-cold fluorescent activated cell sorting (FASC) buffer (Dulbecco's PBS/2% fetal calf serum [FCS]/1% sodium azide) with fluorochrome-conjugated antibodies for 30 min. The following antibodies were applied (Biolegend): anti-CD3 (UCHT1), anti-CD4 (OKT4), anti-CD8 (SK1), anti-CD16 (3G8), anti-CD56 (5.1H11), anti-CD314/NKG2D (1D11), and mouse IgG1κ isotype control (MOPC-21). Cells were washed in FACS buffer prior to acquisition on a BD LSRFortessa™ Flow Cytometer (BD Biosciences) and analyzed with FlowJo, LLC software.

A, CD8+ T-cell receptor and B, CD16 + CD56+ NK cell glycoprotein NKG2D surface expression on flow cytometry histogram with medical fluorophore 1(MF1) quantification. Patient 1 (red) NKG2D surface expression reduced relative to healthy controls (blue) and unstained control (grey). Abbreviations: CD, cluster of differentiation; NK, natural killer; NKG2D, Natural Killer Group 2, Member D; PE, fluorophore R-phycoerythrin.
Results
Immune Studies
Flow cytometry. Our immune re-evaluation with repeat flow cytometry B-cell phenotyping revealed persistent CD5CD19 elevation (93%, Figure 1A). Predominance of naïve B-cells was verified (92%, CD19 + IgD + CD27−; 3.1%, CD19 + CD27 + IgD+; 2.1%, CD19 + CD27 + IgD−; 8.0%, CD19 + CD24++CD38++; Figure 1B). Repeat light chain staining signified polyclonal cells and, hence, the absence of neoplasia (kappa [κ]/lambda [λ] = 62:31). B-regulatory cell (Breg) CD19 + CD24++CD38++ occurrence (8% CD19+ transitional B-cells) was non-aberrant. T lymphocytes subsets (CD4+, CD8+, naïve [CD45RA + CD45RO−], memory [CD45RO + CD45RA−], Tregs [CD127−CD25+], natural killer [NK] cells) were within normal limits. Elevated CD5 B-cell expression (83-97%) was also confirmed in unpublished data on 15 NIAID XMEN patients, which could possibly represent a new biomarker for exploration of XMEN/MAGT1 deficiency.
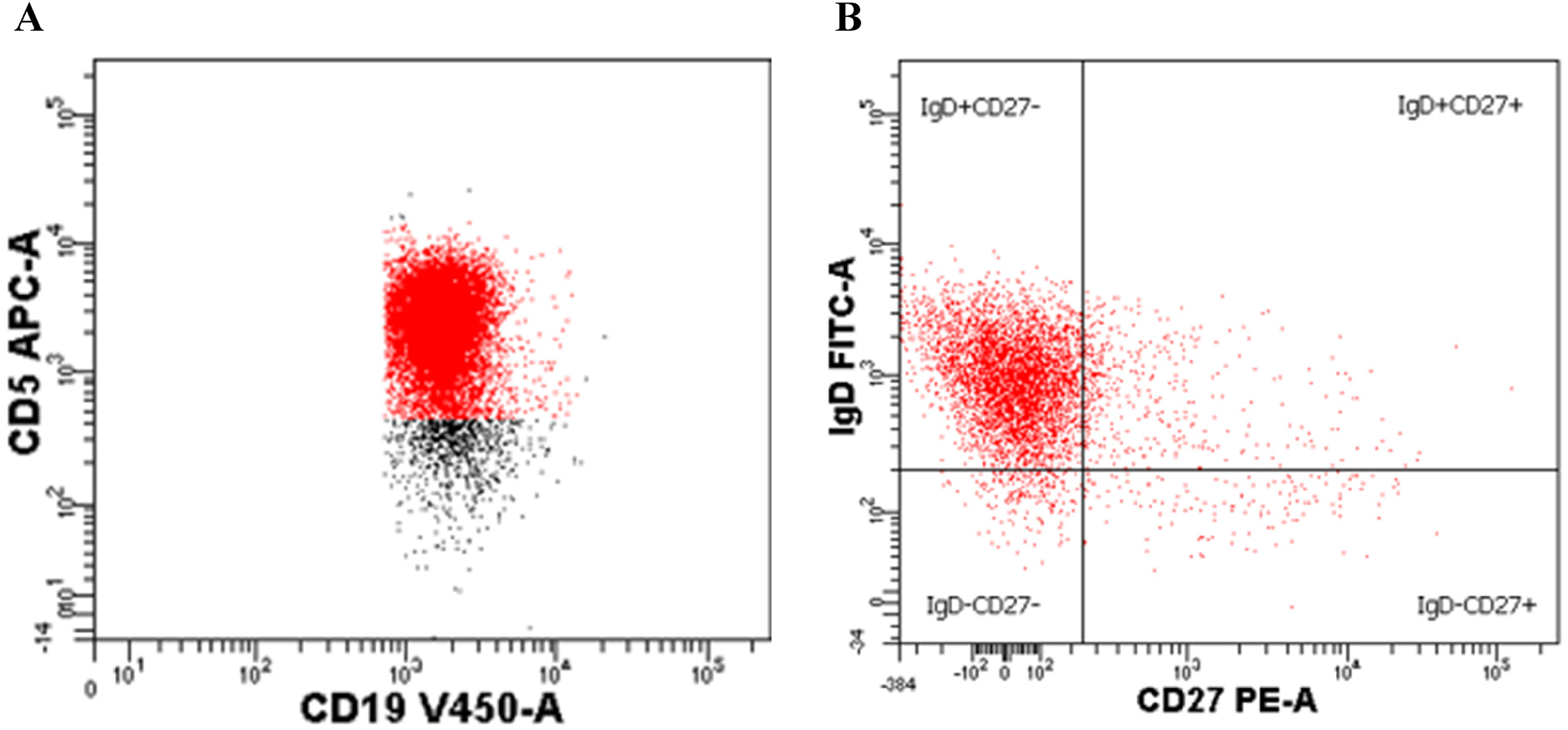
Dual parameter (fluorescence) density plots featuring flow cytometric B-cell phenotyping, targeting specific CD cell surface antigens. A, CD19+-V450- (x-axis) versus CD5+-APC- (y-axis) conjugated monoclonal antibodies indicated CD5+ B-cell (CD5 + CD19+) predominance (93%), quantified by division of CD5 + CD19+ dual-positive cells (right upper quadrant) by total CD19+ cells in the four-quadrant density plot. B, CD 27+-PE- (x-axis) versus IgD-FITC- (y-axis) conjugated antibodies demonstrated single-positive IgD + CD27−CD19+ predominance (97%, left upper quadrant) out of the total CD19+ cells. Antibody conjugate stains APC, V450, PE, and FITC were applied for maximal sensitivity in cell surface antigen analysis (Immune Source, Los Altos, CA) and were conjugated to Becton, Dickinson and Company (BD) HorizonTM V450 Reagents, with 406-nm maximal absorption and 450-nm maximal emission. Abbreviations: CD, cluster of differentiation; APC, allophycocyanin; A, conjugated monoclonal antibody; V450, violet laser 450/50-nm absorption/emission reagent; PE, phycoerythrin; Ig, immunoglobulin; FITC, fluorescein isothiocyanate.
ΦX174 bacteriophage antigen immunization. Dual ΦX174 bacteriophage immunization first demonstrated a diminishing primary immunization response with minimal elevation, within normal range below the average control population antibody response (Figure 2A). The secondary immunization response declined without elevation to two standard deviations below normal control range (Figure 2B). Abolished antibody activity after 2-mercaptoethanol A exposure indicated IgM predominance.12,13

A and B, Primary and secondary antibody responses to phiX174 (ΦX174) bacteriophage antigen immunization in Patient 1. Blood samples of antibodies were measured for rate of neutralization (Kv, K value) in weekly intervals. Secondary immunization was administered 6 weeks after primary immunization. Normal ranges present antibody titers in a control population antibody response. Abbreviations: Kv, rate of antibody neutralization; PID, primary immunodeficiency.
Next-generation sequencing. Genomic analysis revealed a likely pathogenic c.923-1_934 deletion (splice acceptor), loss of function (LOF) mutation in the magnesium transporter 1 (MAGT1) gene hemizygous splice site gene, classified under combined immunodeficiency, autoimmune lymphoproliferative disorders, EBV-associated lymphoproliferative syndromes, monogenic autoimmunity and autoinflammatory disorders, with reference to MAGT1 immunological signaling roles studied by Wolf and Trapani.14,15 The increased risk allele c.2104C > T (p.Arg702Trp), associated with increased risk of Crohn's disease, was identified in the NOD2 gene linked to autosomal dominant Blau syndrome. Variants of uncertain significance identified in PARN (c.34C > T [p.Leu12Phe]), PRKDC (c.4375C > T [p.His1459Tyr]), and ZAP70 (c.1415G > A [P.Arg472Gln]) genes. This NGS has since expanded to associate MAGT1 with congenital disorders of glycosylation and hereditary hemophagocytic lymphohistiocytosis (HLH) disorders panels, as well as metabolic disorders newborn screening.
Flow cytometric histogram with medical fluorophore 1 quantification. NKG2D expression plotted on flow cytometry histograms with MF1 quantification was reduced in Patient 1, compared to healthy controls, in both CD8+ T cells and CD16 + CD56+ NK cells (Figure 3).
Treatment
Intravenous immunoglobulin (IVIG) was not indicated, due to the lack of infectious severity and frequency. The patient continues with annual follow-up.
Discussion
PIDs are heterogenous inborn errors of adaptive or innate immunity implicated in nearly 2% of the coded human genome,1,2 resulting in increased susceptibility to infection, autoimmune disease, inflammation, and malignancy. 1 Initial immunological evaluation is guided by clinical manifestations, as well as exclusion of immunosuppressive therapy or other acquired causes of secondary immunodeficiency. 1 Laboratory screening of immune function for humoral immunity, as initially evaluated in Patient 1, includes serum Ig levels, specific antibody titers, antibody response to booster immunization, and flow cytometry to enumerate total B-cells. 1 Patient 1's further immunological evaluation follows advanced laboratory test recommendations for flow cytometry of B-cell subsets (Figure 1), in vitro Ig production in response to mitogens or other stimuli, and antibody response to immunization with ΦX174 (Figure 2). 1 His atypical unequivocal diagnosis, prognosis, and treatment indicated additional PID definition at the molecular genetic level, 1 which identified his MAGT1 mutation and, subsequently, enabled genotypic-phenotypic association with XMEN.
Many clinical similarities were identified between PID cohort and features of XMEN. Patients in the PID cohort were experiencing recurrent and frequent episodes of otitis media, sinusitis and pneumonia since a young age. This is the most common clinical finding in XMEN disease. Other clinical findings included an acute onset hemiplegia at age of 2 in patient 1 that resolved slowly over a period of 3-4 months, it can be compared with possible neurodegeneration and other neurologic finding like Guillan Barre Syndrome seen in XMEN. Another common feature between the 2 is autoimmune disorders, in the PID cohort Alopecia totalis was common and patient 1 was diagnosed with autoimmune hepatitis.3,8
The updated International Union of Immunological Societies (IUIS) Expert Committee on Inborn Errors of Immunity (IEI) classifies XMEN with the MAGT1 genetic defect under “Diseases of immune dysregulation” (IV), then “Susceptibility to EBV” (b). 2 Specified characteristics include EBV infection, lymphoma, viral infections, respiratory and gastrointestinal infections; glycosylation disorder; and neurological manifestations as clinical features. 2 Immunological factors include low CD4, low recent thymic emigrant cells, poor proliferation to CD3, with 2019 updates of high B cells without inclusion of CD5 expression and dominant negative T cells. 2 This XMEN disease definition is followed by protein kinase, DNA-activated, catalytic subunit (PRKCD) deficiency with comparable manifestations to XMEN: recurrent infection, EBV chronic infection, lymphoproliferation, and systemic lupus erythematosus-like autoimmunity (nephrotic and antiphospholipid syndromes). 2 No T-cell deficits, IgG hypogammaglobulinemia, and low memory B cells are specified immunological defects of PRKCD deficiency. 2 High CD5 B-cells specification under PRKCD deficiency shares B-cell predominance with Patient 1, but MAGT1 and protein kinase C, δ (PRKCD) mutations, rather than PRKDC, were distinguished on NGS. 2
CD5+ PID is characterized by an abnormal preponderance of naïve CD5 B-cells.3–6 All CD5+ PID patients diagnosed before accessible genetic testing demonstrate consistency in male sex, 3 CD5 B-cell predominance, and SPAD with poor antibody response to polysaccharide and bacteriophage vaccination. 3 Phenotypes and severity of illness are variable.3–6 Several demonstrated pan- or partial hypogammaglobulinemia requiring IVIG. Autoimmune manifestations, most commonly alopecia totalis, were diagnosed in five of the patients, including Patient 1 with autoimmune hepatitis.3–6 MAGT1 deficiency is only specified in Patient 1 (c.923-1_934 del), as the other patients did not undergo further diagnostic evaluation at the molecular genetic level due to patient availability.
Unearthing the MAGT1 LOF mutation role in XMEN disease has progressed from initial nonspecific association of reduced intracellular Mg2+ with T-cell receptor (TCR) signaling dysregulation, resolvable with Mg2+ supplementation.14–17 Gotru et al investigated this proposed cation signaling defect leading to immune dysregulation with a MAGT1-deficient mouse line (Magt1/y). 18 Mg2+ homeostatic disruption in Magt1/y B cells and increased calcium influx after B-cell receptor (BCR) stimulation in vitro contraindicated earlier suggestions of dysregulated T-cell effector predominance. 18 Proliferative CD19+ and marginal zone B-cells and reduced plasma B-cells among CD45+ splenocytes in vivo support earlier investigations of specific B-cell subset immune-modulatory roles,19,20 the CD5 PID cohort B-cell dysregulation, as well as the B-cell predominance tested in other XMEN patients (83%-97%). In addition, although a definitive mechanism has not been identified, in a publication by Watson et al, B cells were predominantly of a naive type in the XMEN phenotype, which would correlate with CD5 expression on B cells. 21
MAGT1 is now confirmed as a non-catalytic subunit of the OST and facilitator of asparagine (N)-linked glycosylation (NLG) of specific substrates. 8 This 10-exon gene located on chromosome Xq21.1 is speculated to contain multiple in-frame translation initiation sites encoding a 335 amino acid protein (UniProtKB-Q9H0U3). 8 This sequence change affects an acceptor splice site in intron 8 of the MAGT1 gene. It is expected to disrupt the RNA splicing and likely results in an absent or disrupted protein product. This variant is not present in any population databases and has not been reported in the literature in individuals with MAGT1-related disease. Most deleterious MAGT1 molecular alterations, such as the c.923-1_934del LOF mutation identified in Patient 1, have undetectable protein expression on immunoblotting. 8 Donor and acceptor splice site variants typically lead to a loss of protein function, 22 and loss-of-function variants in MAGT1 are known to be pathogenic. 16 The “N-linked glycosylation defect” addition to the revised XMEN disease title proposed by Ravell et al acknowledges this recent glycobiological research classification of MAGT1 deficiency as a selective congenital disorder of glycosylation (CDG). 8 This impaired glycosylation predominantly affects the immune system through decreased expression of NKG2D. This activating receptor is critical for cytotoxic function against EBV, here it is poorly glycosylated and invariably decreased on CD8+ T Cells and Natural Killer cells from XMEN patients; it is the best biomarker of the disease.8,9 Dr Ravell, of the NIH who described the above findings and initially described the XMEN disorder with MAGT1 mutation, examined the patient's blood further confirming our patient's phenotype. Serological evidence of EBV-positive lymphoproliferative disease has not been detected in approximately half of diagnosed XMEN and our CD5+ PID cohort, regardless of reduced NKG2D expression. 9 In addition, Ravell's study showed 27 of 36 patients with XMEN had recurrent infections, 23 out of 30 had hypogammaglobulinemia, 25 out of 34 had EBV viremia, and 4 out of 36 had. VZV. Unpublished data from Dr Ravell stated that all XMEN patients had similar CD19 and CD5 expression. The variable phenotypic presentations of diagnosed XMEN patients and Patient 1 are consistent with their diverse MAGT1 mutation variations,8–10,23–28 which we suspect in the remaining CD5+ PID cohort which did not have genetic testing available. The patient that has been described was one of the two original patients described with CD5+ PID. The other patient is deceased; and other patients have been lost to follow-up in other cities and countries.
Several case reports and reviews since this in vitro evidence of B-cell predominance by Gotru et al have prompted recent total B-cell phenotyping in known XMEN patients, including 95% of the 23 patients studied by Ravell et al 9 However, thorough B-cell subset phenotyping had not been published for XMEN patients, apart from Patient 1. Two populations of naive B cells (CD20 + CD27−CD22 + IgM + HLA−DR + CXCR5 + CXCR4++CD10 + CD38+ and CD20 + CD27−CD22 + IgM + HLA−DR + CXCR5 + CXCR4 + CD10−CD38−) were found to distinguish XMEN, autoimmune lymphoproliferative syndrome (ALPS), and healthy individuals. 9 The gold diagnostic standard of reduced NKG2D expression is documented for most XMEN patients (95-100%), 9 in addition to Patient 1 (Figure 3), and has been the target of recent messenger RNA electroporation-corrected autologous T and NK cells for potential short-term cell therapy. 29 This reduced NKG2D expression is the only T-cell abnormality identified in Patient 1, suggesting a preponderance of B-cell dominance in this disease spectrum. Other notable lab findings include transient elevation of hepatic enzymes,8,9 consistent with autoimmune hepatitis and ulcerative colitis in Patient 1.
Multiple case reports and reviews reveal heterogenous phenotypes of diagnosed XMEN patients. Recurrent sinopulmonary infections, chronic lymphadenopathy, splenomegaly, lymphoma, EBV-negative malignancy, and various autoimmune conditions are among clinical manifestations.8,9 Patient 1 presented with recurrent viral infections and autoimmune symptoms, although the severity and frequency are mild compared to some XMEN patients. He also lacks evidence of longstanding EBV and subsequent neoplasia. We speculate that his unique phenotype is closely associated with the B-cell abnormalities studied by Gotru et al 18 Such disturbance of altered BCR signaling by MAGT1 deficiency may translate to the proliferative CD5 expression in MAGT1 deficiency/CD5+ PID as a biomarker of B-cell maturational arrest, resulting in aberrant response to polysaccharide antigens and autoimmune manifestations.
The major limitation to the study is that it involved only one of the CD5 PID patients. The described was one of the two originally reported cases. The other patients passed away. Other CD5 PID patients were lost to follow up in other states and other countries.
History of recurrent infection and verification of MAGT1 mutation and reduced NKG2D expression in Patient 1 support our relation of CD5+ PID and XMEN as identical diseases or subset of the other. Advanced literature on XMEN has indicated broader phenotypic and genotypic variance than the “XMEN” title pathologies.8–11 The recent proposition to update the acronym to “X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect” acknowledges recent confirmation of the MAGTI mutation causation of glycosylation disorder and acknowledge of B-cell abnormalities.8,9 Further B-cell subset phenotyping in presently diagnosed patients with identified MAGT1 mutation and/or diagnosed XMEN, external to the reported cases of this authorship,3–6 is warranted to verify CD5 B-cell predominant expression prevalence. We demonstrate consistencies of CD5+ PID with XMEN to be acknowledged among that spectrum of MAGT1 deficient patients and considered in future classification refinements in the IUIS Expert Committee of IEI. 2
Conclusions
The major conclusions that can be drawn from this study is that there are consistencies of CD5+ PID with XMEN, and that this disease that is illustrated within this manuscript should be acknowledged within the spectrum of MAGT1 deficiency and considered in further refinements of classifications of this range of disease.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contribution(s)
Ackowledgment
We acknowledge and are grateful for the work of Dr Ravell of the NIH in his performance of key NKG2D flow cytometry analysis and detailed information on XMEN.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.