
Abstract
Acalvaria is a rare congenital malformation of unknown etiopathogenesis that affects the development of the skull and surrounding muscular structures. There is scant literature regarding the pathophysiology, and no standardized guidelines exist for patient management. Herein, we present a case of acalvaria in a newborn and our multidisciplinary approach from diagnosis to 1 year of age.
Background
Acalvaria is a congenital malformation characterized by the absence of the flat bones of the skull, dura mater, and associated muscles in the presence of normal cranial contents and facial bones.1-3 Acalvaria has been associated with additional anomalies including holoprosencephaly, micropolygyria, facial clefts, cardiac anomalies, and club foot.1,2 The embryologic development is not well understood, and some hypothesize that there is a post-neurulation defect resulting from failure of migration or differentiation of tissue.1-5 Acalvaria has previously been considered a fatal diagnosis, with ability to diagnose prenatally, with subsequent elective termination.1-3,6
Due to the scarcity of cases and lacking literature, there are no standardized guidelines for patient management. Supportive care and management of associated anomalies are the current recommendations.1,2 However, 1 case attempted skull reconstruction with bone grafting and cranioplasty at birth without complications at the 22-month check-up. 7 Conversely, other reports suggest there has been spontaneous bone growth following initial conservative management. 1
Herein, we present a postnatal diagnosis of acalvaria in a term, female infant with abnormal exam findings requiring facility transfer for additional diagnostic evaluation. Prenatal ultrasounds of the infant were reportedly normal. CT Head after birth demonstrated absence of the bilateral parietal, occipital and superior frontal calvarium.
Case Report
A female infant was born to a 21-year-old G3P2 mother at 38 weeks and 1 day gestation via spontaneous vaginal delivery. Maternal history was significant for chronic hypertension, obesity, HPV positive, and chlamydial infection during pregnancy. No other complications were appreciated throughout pregnancy, and there were no maternal medications outside of antibiotics. Routine neonatal resuscitation was provided and APGARs were 8 and 9 at 1 and 5 minutes, respectively. Initial physical examination at an outside institution was significant for bogginess on palpation of the head, concerning for enlarged fontanelles. Otherwise, the physical examination was normal without neurologic deficits.
Skull series (Figure 1) and brain ultrasound were obtained, demonstrating abnormal calcification of the calvarium, but no acute concerns on ultrasound. The infant was feeding, voiding, stooling, and moving all 4 extremities appropriately. Transfer was arranged due to the need for additional advanced imaging and subspecialty evaluation.
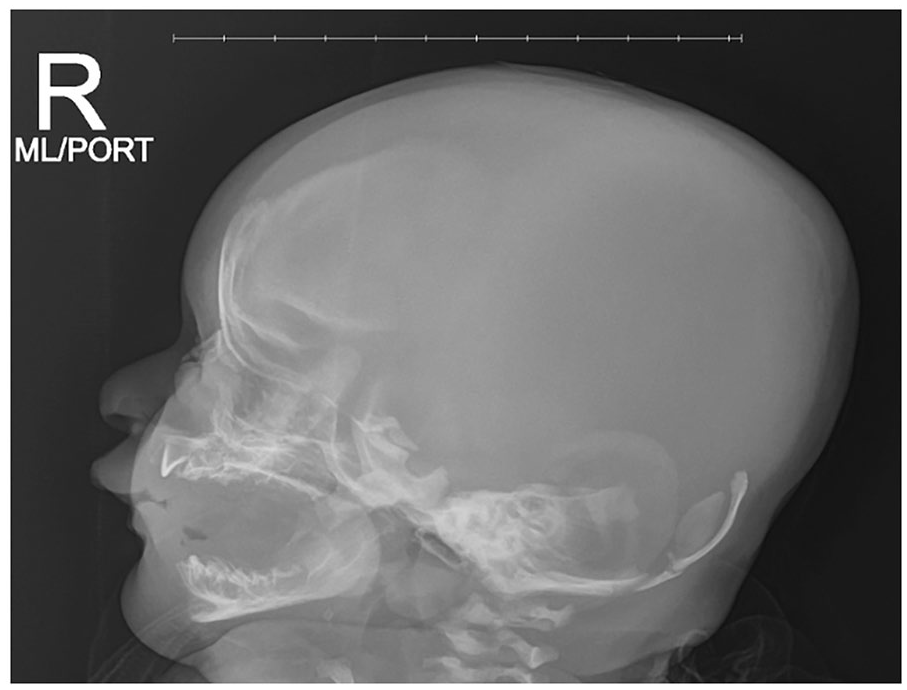
Skull Series. Sagittal view. Day of life 1. Demonstrating decreased mineralization of the calvarium with abnormally enlarged anterior fontanelle.
Upon arrival to our institution, the infant had a normal neurologic and physical examination aside from the absence of cranial bones. Initial precautions included a foam donut cushion and frequent neurologic monitoring. CT Head without contrast with 3D reconstruction (Figure 2) was obtained, confirming the absence of bilateral parietal, occipital, and superior frontal calvarium. Initial consults included pediatric neurosurgery, plastic surgery, and genetics. Additional workup included a skeletal survey, abdominal ultrasound, and echocardiogram. All additional imaging was negative, except for the incidental finding of an interrupted IVC. An Invitae Skeletal Disorders Panel was sent, later revealing a DMRT2 variant of uncertain significance (VUS).
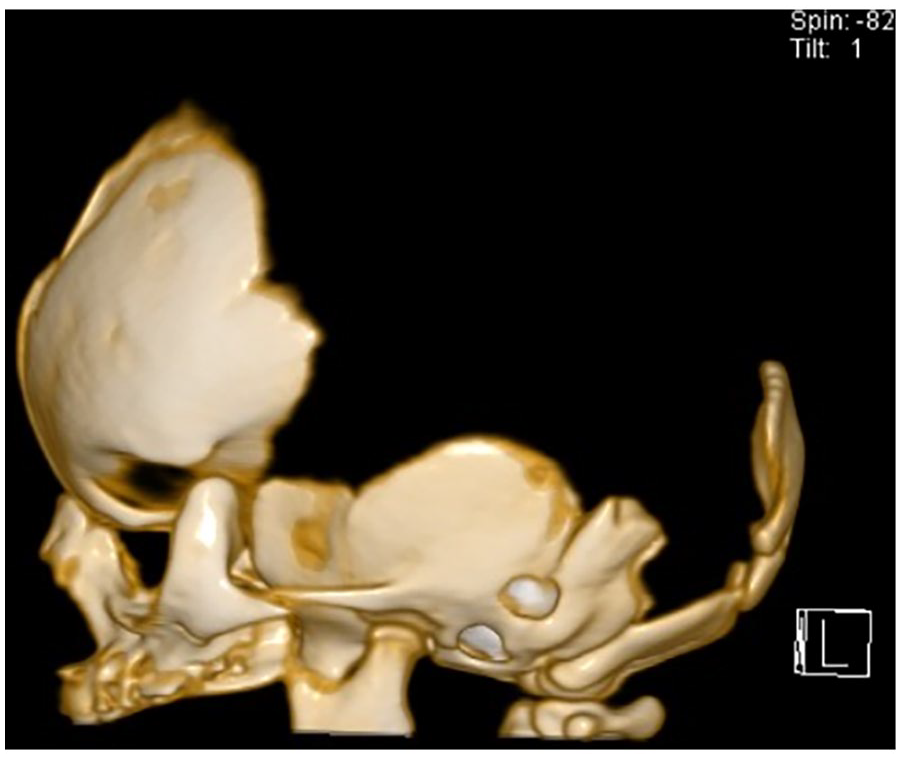
CT Head with 3D Reconstruction. Sagittal view. Day of life 1. Demonstrating absence of bilateral parietal, occipital, superior frontal calvarium.
Pediatric nephrology and pediatric endocrinology were consulted given transient hyperphosphatemia. PTHrP and ALP levels were normal and Vitamin D 25-OH levels insufficient (20 ng/mL) with subsequent supplementation. Variability in phosphorus is believed to be related to acalvaria and possible metabolic bone disease.
The patient was discharged on day of life 8 with outpatient management consisting of protecting the cranium with foam padding when supine. Follow-up with pediatric neurosurgery, plastic surgery, and genetics scheduled.
Discussion
We report a rare case of acalvaria in an otherwise healthy newborn infant, with reportedly normal prenatal ultrasound findings. At the time of discharge, the neonate was neurologically intact and feeding, voiding, and stooling without issues.
There are variations in the presentation of acalvaria. Many of the recorded acalvaria cases are electively terminated before birth. Four living cases are either living with severe neurological deficits,1,7 or are associated with other severe abnormalities including cutis aplasia1,8 orbit and eyelid defects, 7 or are without associated abnormalities. 1 Our case is unique in that there were no abnormalities detected on prenatal ultrasounds, nor were there any neurological deficits present at birth, which may be the second known case.
The patient has been unable to obtain follow-up with genetics in person due to a variety of reasons. However, via telemedicine, genetics noted that the DMRT2 VUS has no well-established disease association, except some evidence of correlation with autosomal recessive spondylocostal dysostosis. 9 Given the patient only has acalvaria and the skeletal survey was otherwise unremarkable, there is low concern that this VUS is contributing to the calvarial abnormality.
At the initial neurosurgery follow-up, a new small island of bone growth above the right ear was appreciated. The plan at that time was to consider helmet administration with the idea that pressure would stimulate growth, though with insufficient evidence at this time. Ophthalmology evaluated prior to helmet administration to rule out retinal anomalies or optic disk edema. However, at the next follow-up, the plan was to monitor conservatively given repeat CT Head findings at 8 months of life with increased bone growth appreciated (Figure 3) without the use of a helmet. At 1 year follow-up, the team continued with conservative management, only considering a helmet for safety purposes, now that patient has begun walking. X-rays at 12 months of life demonstrated further mineralization (Figure 4).
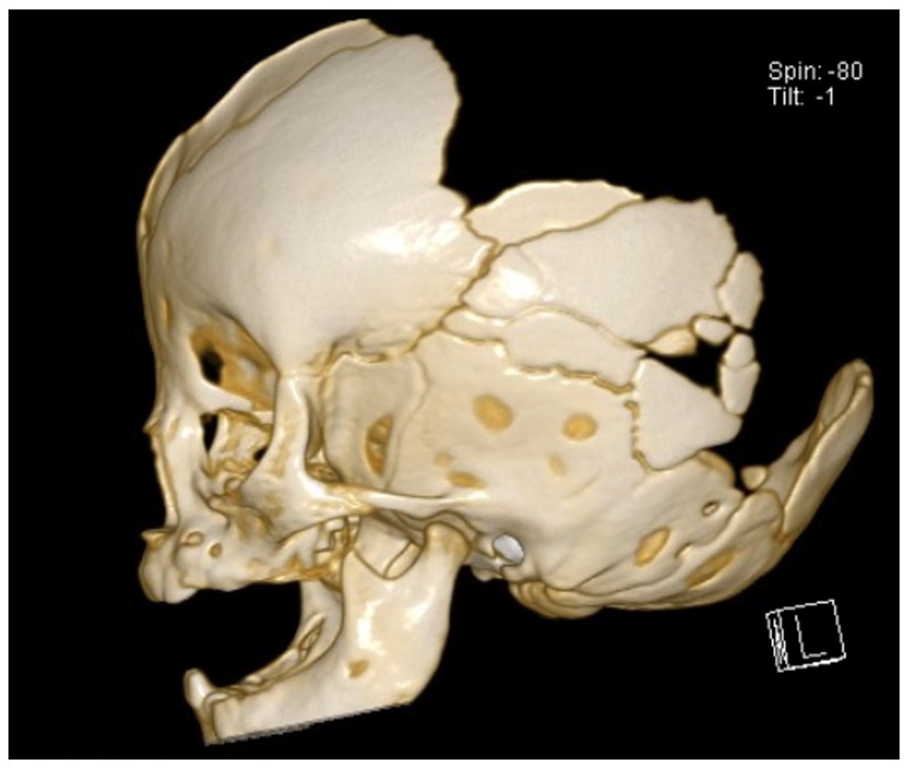
CT Head with 3D Reconstruction. Eight months of life. Sagittal view. Demonstrating absence of bilateral parietal, occipital, superior frontal calvarium with increased calcification.
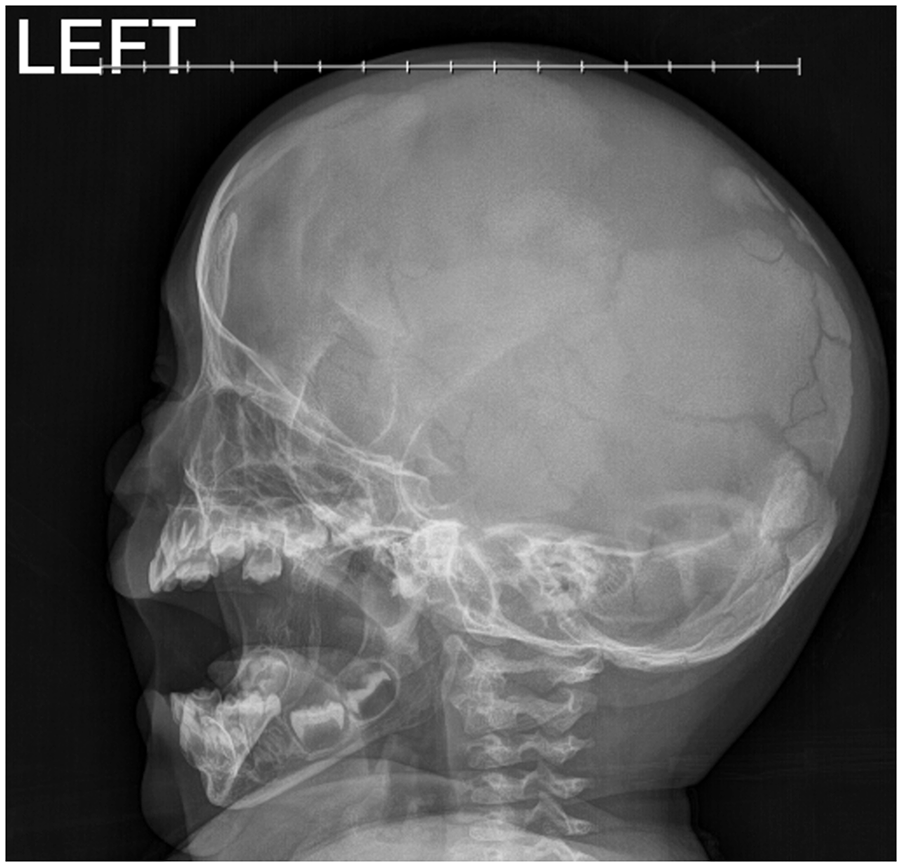
Skull Series. Sagittal view. Twelve months of life. Demonstrating increased mineralization of the calvarium.
At initial plastic surgery follow-up, imaging was reviewed with a plan for conservative management and repeat CT every 6 months to evaluate bone growth. If ossification was not complete by age 1, would consider protective helmet until old enough to undergo alloplastic cranioplasty. Of note, patient was not a candidate for autologous bone grafting due to large defect size and lack of sufficient donor site. At 1 year follow-up, the patient was noted to be meeting all developmental milestones with improvement in overall bony growth (Figures 5 and 6). The plan was to repeat imaging in a few months, continue with conservative management, and likely consider bony reconstruction with porous polyethylene implant around age 4 to 5 years.
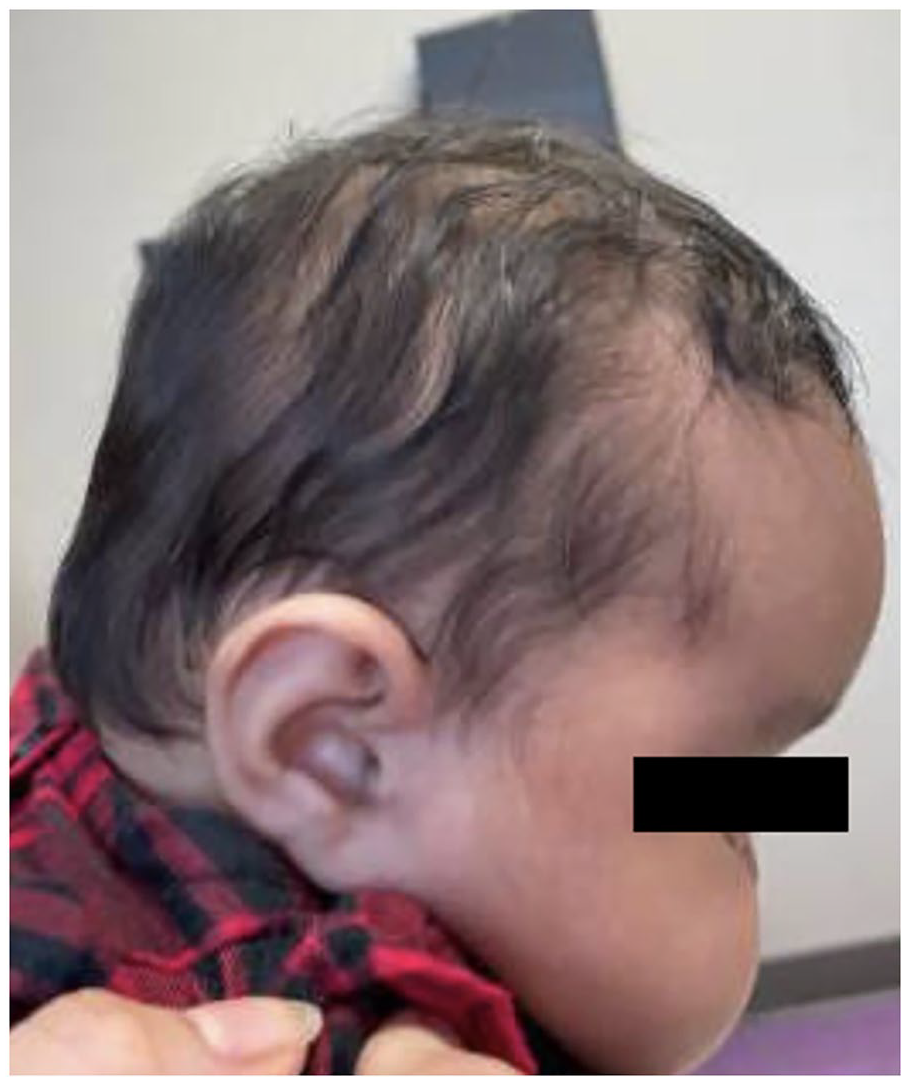
Phenotype. Sagittal view. First few months of life.
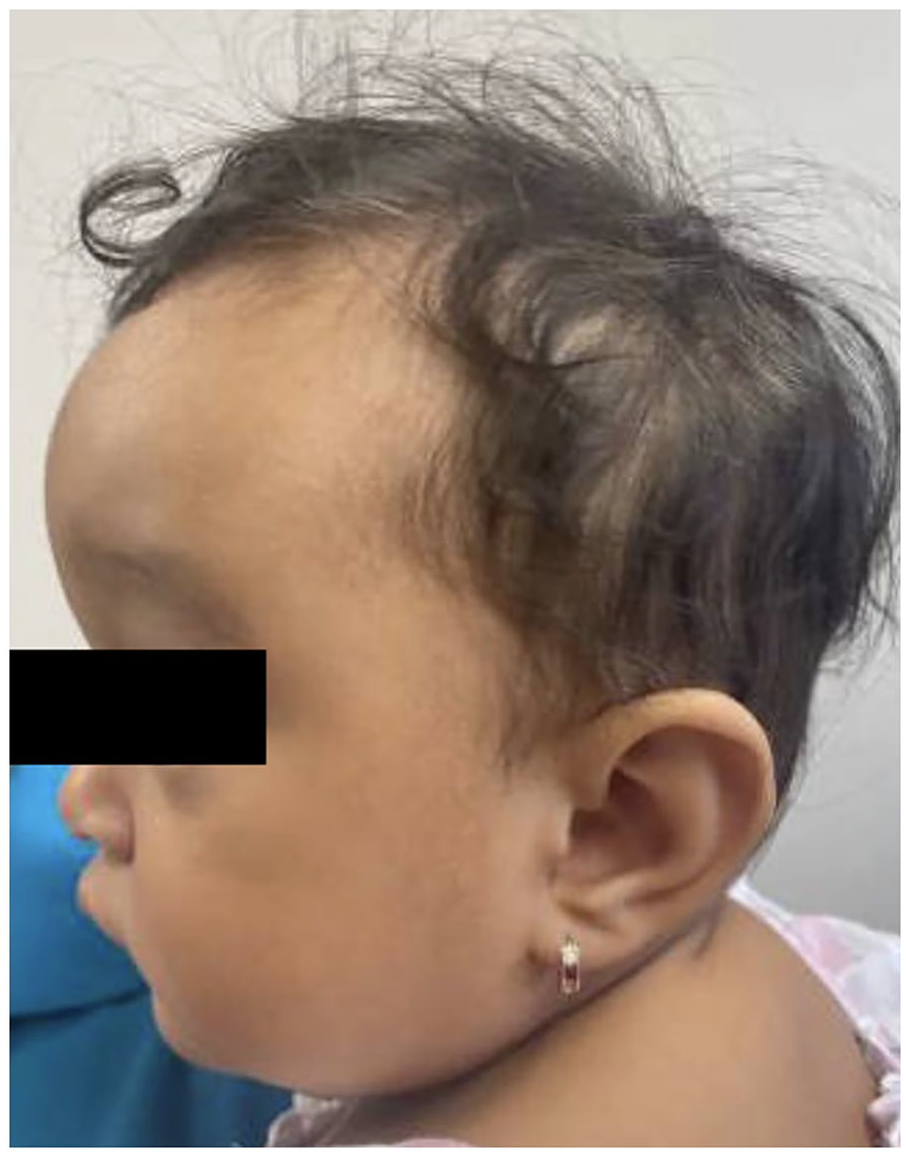
Phenotype. Sagittal view. Nine months of life.
Overall, in this unique case of isolated acalvaria, multiple disciplines recommended conservative management with close monitoring of developmental milestones and bone development. Obtaining a skeletal survey, Invitae Skeletal Disorders Panel, along with general labs related to bone development upon birth were recommended to rule out other concerns. Parental education around neuroprotection was vital. Helmets were considered for various reasons, with the latest as a form of protection given patient ambulation. There is ongoing consideration of bony reconstruction in the near future. However, it is important to note that management likely would have been different if other abnormalities were present, as with the aforementioned cases.
This case is critical to report to the scientific community as it confirms the speculation that there are more mild cases of acalvaria with the potential to develop normally. This could change the current management of elective termination before birth or include more progressive treatment, such as surgery, to improve cosmetic outcome of the skull over time. The limitation of this case is that there needs to be longer follow-up and tracking of this patient’s progress throughout their life.
Footnotes
Ethical Approval
Institutional Review Board approval was not required.
Informed Consent
Consent was obtained from the patient’s parent.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.