
Abstract
Cleidocranial dysplasia (CCD) and Alexander disease (AxD) are rare, autosomal dominant disorders that are characterized by a mutation in the runt-related transcription factor 2 (RUNX2) and the glial fibrillary acidic protein (GFAP) genes, respectively. There is no known relationship between RUNX2 and GFAP which would cause co-morbidity. We report a rare case of a 13-year-old with CCD who came to the clinic complaining of a 2-year history of progressively worsening episodic exacerbations of bulbar, ataxia, nystagmus, kyphoscoliosis, and nausea, but was intact cognitively. Initial diagnosis was a difficult process because preliminary symptoms for Juvenile AxD overlapped with previously diagnosed CCD and the initial genetic test identified our patient’s GFAP gene as a variant of uncertain significance. Our experience emphasizes the importance of continuing to report pathogenic variants of GFAP for AxD to build on our existing compendium of variants for GFAP for quicker and more efficient diagnosis of AxD.
Introduction
Cleidocranial dysplasia (CCD) is an autosomal dominant skeletal disease that has varying phenotypes, characterized by inherited or de novo mutations in the runt-related transcription factor 2 (RUNX2) gene. 1 Patients with RUNX2 gene mutations often present with open or delayed closure of calvarial sutures, hypoplastic or aplastic clavicles, dentition anomalies, and supernumerary teeth. 2 Though early recognition is essential for successful therapy and treatment, there is wide variability in the presentation of skeletal and extraosseous symptoms and many cases are misdiagnosed. Alexander disease (AxD) is a rare, autosomal dominant neurodegenerative disorder that is caused by a pathogenic gain-of-function variant in the glial fibrillary acidic protein (GFAP) gene. 3 AxD is predominantly seen in patients <2 years of age. It is a leukodystrophy characterized by the accumulation of GFAP-rich proteins in astrocytes, which cause the characteristic formation of Rosenthal fibers (RFs) in the subpial, perivascular, and subependymal regions of the cortex and white matter. 4 These have also been recently linked to aberrant Ca2+ responses in astrocytes. 5 Current research also suggests that genetic anticipation may occur in familial AxD and genetic mosaicism could explain this phenomenon. 6 We examine a 13-year old male with confirmed genetic diagnoses of both CCD and AxD. To date, there is no known relationship between RUNX2 and GFAP which would cause co-morbidity of both disorders. However, in one study of cultured neocortical astrocytes, RUNX2 protein was markedly expressed in cells expressing GFAP, with selective localization in the nucleus. 7 The relationship remains unclear.
Case Report
A 13-year-old with cleidocranial dysplasia, vitamin D deficiency, seasonal allergies, and lab-negative celiac disease. The patient was diagnosed clinically with cleidocranial dysplasia at birth with absent clavicles, mild macrocephaly, mild hypertelorism, and a high-arched palate. At this time, the CCD diagnosis was not genetically confirmed. Initially, the patient presented with 2 years of episodic dizziness, nausea, and choking. The initial frequency was every 4 to 6 months but progressively increased to every 2 to 4 months. The duration of episodes ranged from hours to 3 to 4 days. At baseline, he was not able to lay flat on his back since early childhood. The neurological examination was notable for mild dysarthria and dysphonia, originally thought to be baseline from his CCD, directional changing nystagmus, kyphoscoliosis, hyporeflexia, and a wide-based gait. An MRI of the brain without and with contrast found extensive nodular signal abnormality and enhancement involving the pons, medulla, and bilateral middle cerebellar peduncles (see Figure 1). Extensive confluent signal abnormality involved the periventricular white matter and bilateral basal ganglia. These results prompted an extensive workup for neuroimmunological, oncological, rheumatologic, infectious, nutritional, and genetic etiology. Cerebellar biopsy consisted of neutrophils with extensive Rosenthal fibers, reactive gliosis, eosinophilic granular bodies, and focal perivascular lymphocytic cuffing. There was no mitotic activity or necrosis identified. The presence of Rosenthal fibers prompted the treatment team to offer genetic testing through a next-generation sequencing panel for leukodystrophies and leukoencephalopathy at a commercial laboratory, which tested 446 genes. The parents also agreed to concurrently confirm the CCD diagnosis through genetic testing. Results confirmed that the patient had CCD with a RUNX2Exon 4 c.577C>T (p.Arg193
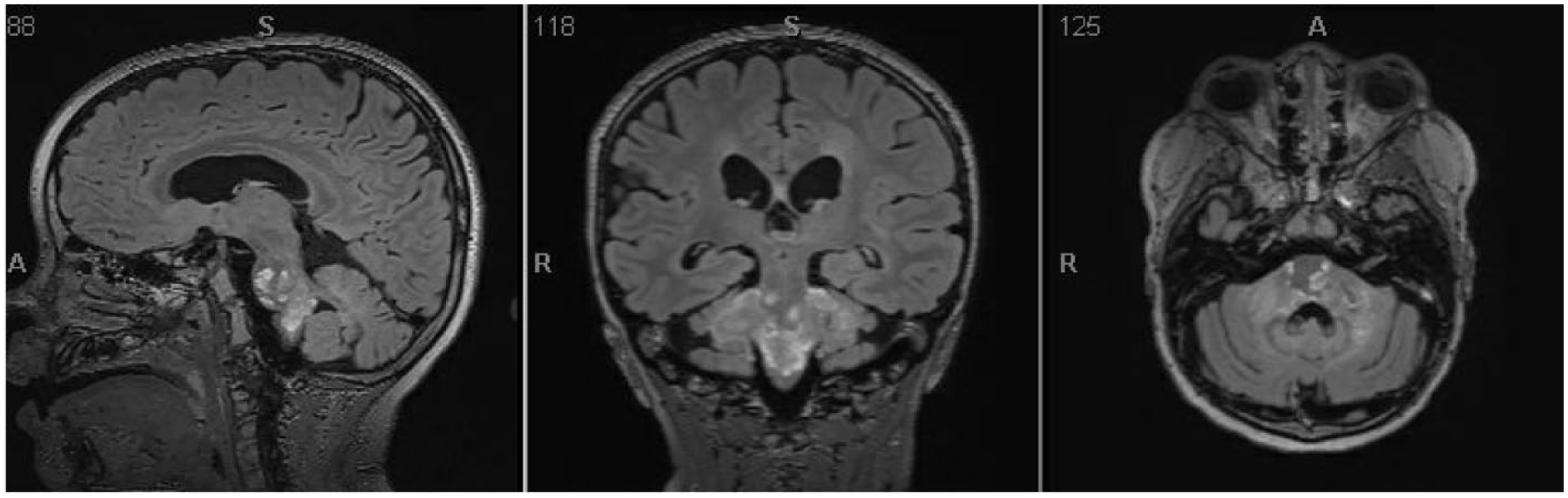
T2-weighted axial MRI sections of the patient’s brain show extensive nodular signal abnormalities and enhancement involving the pons, medulla, and bilateral middle cerebellar peduncles. There is also extensive confluent signal abnormalities involving the periventricular matter and bilateral basal ganglia.
Discussion
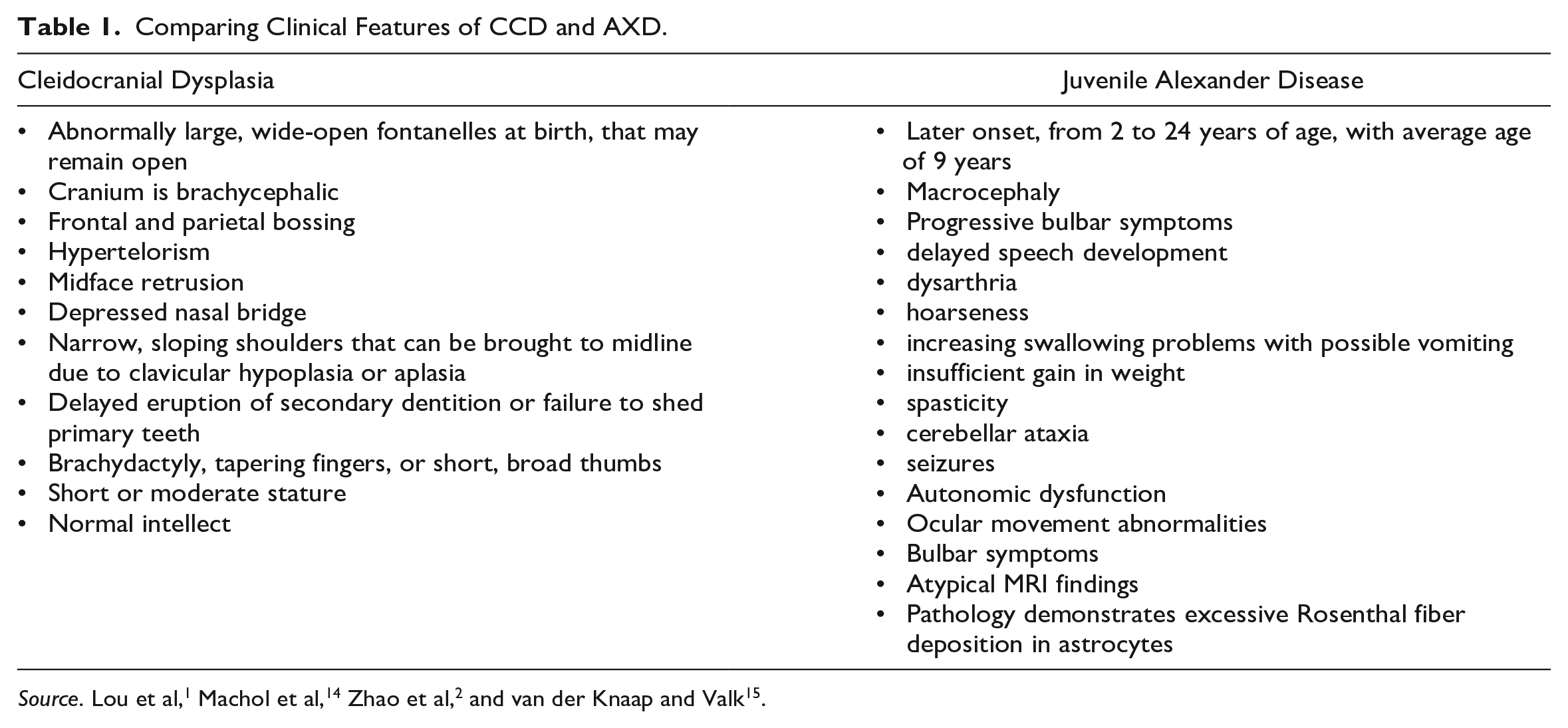
By themselves, CCD and juvenile AxD are rare genetic disorders. In this case, overlapping features of scoliosis, mild dysarthria, and dysphonia made the diagnosis even more elusive. A brief review of the literature showed that the co-occurrence of 2 genetic disorders for skeletal deformities is well documented. 11 For example, Liu et al 11 found that patients with dual molecular diagnosis presented blended phenotypes of the 2 diseases, which greatly benefited from exome sequencing. Günthner et al 12 presented a case that used whole exome sequencing to serve as a powerful instrument to determine 2 independent genetic pathologies. Balci et al 13 found that consanguinity and multisystem disease increased the likelihood of incidence of multiple genetic diagnoses in a family. Because genetics testing is a growing field, our recommendation is that diagnosis of AxD and CCD be based on clinical presentation, versus determined strictly by genetic testing. Craniofacial surgeons should have a general familiarity with the signs, symptoms, and history of a patient’s genetic disorder, prompting further evaluation for second diagnoses if the presentation of symptoms is atypical (see Table 1). Our experience emphasizes the importance of continuing to report pathogenic variants of GFAP for AxD to build on our existing compendium of variants for GFAP for quicker and more efficient diagnosis of AxD.
Comparing Clinical Features of CCD and AXD.
Footnotes
Ethical Approval
This case report was done in accordance with the Declaration of Helsinki. The collection and evaluation of all protected patient health information was performed in a Health Insurance Portability and Accountability Act (HIPPA)-compliant manner. Informed consent was obtained prior to publication of all photogram and images included herein.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.