
Abstract
Background:
Temperate bacteriophages are widespread in bacterial genomes and can play significant roles in bacterial evolution and pathogenicity. Despite their importance, they remain poorly characterized in nonclinical Staphylococcaceae, particularly Mammaliicoccus sciuri.
Materials and Methods:
We analyzed 26 M. sciuri strains isolated from the nasal cavities of East African dogs and camels. Prophages were induced using mitomycin C, and isolated phages were characterized by whole-genome sequencing, phylogenetic analysis, electron microscopy imaging, and host-range determination.
Results:
Eight novel siphoviruses were isolated. Phylogenomic analysis revealed two new families, each comprising two genera. Notably, phages from one of these families (with genomes >130 kbp) exhibit a broad host range, while the other family is related to previously described phages implicated in horizontal gene transfer.
Conclusion:
Our findings reveal unexpected diversity of temperate phages in M. sciuri, expanding current knowledge of phage distribution in animal-associated opportunistic pathogens.
Introduction
Prophages play a crucial role in Staphylococcaceae by contributing to horizontal gene transfer (HGT) and genetic diversity. Together with other mobile elements such as plasmids, they contribute to the genetic diversity of bacteria between pathogenic bacteria and commensal bacteria.1,2 In Staphylococcus aureus, prophages can carry genes encoding virulence factors such as Panton-Valentine leukocidin, staphylokinase, or enterotoxins, thus contributing to virulence and pathogenicity.3,4 In Mammaliicoccus sciuri, prophages facilitate HGT, potentially influencing its adaptation and interaction with other bacteria and the host.5,6 For Staphylococcaceae, most reported temperate phages have been isolated from clinical isolates of S. aureus, which does not reflect the full spectrum of diversity of staphylococcal phages. A recent study showed that staphylococcal phages isolated from nonclinical samples appeared to have a wider host range than previously reported. 7 Most of the phages were able to infect several Staphylococcus species, even if they tended to predominantly infect more strains of one particular species, indicating they could facilitate HGT between different species. Similarly, staphylococcal phages with larger genomes (∼145 kbp), including species in the genera Silviavirus 8 and Kayvirus, 9 are also known to have extended host ranges that might facilitate the HGT between different bacterial species 10 and could carry large genetic elements such as SCCmec elements or genetic islands, which can be mobilized by phages. 6 These recent discoveries encourage further investigation of the staphylococcal phage diversity employing isolates that mark novel phylogenetic lineages or that have been isolated from specific ecological niches to better understand their diversity and roles.
M. sciuri (previously known as Staphylococcus sciuri) was first described as a Gram-positive, coagulase-negative commensal bacterium of wild and domestic animals, humans, and environments. 11 It was later discovered that M. sciuri can also act as an opportunistic pathogen, capable of causing severe clinical diseases in a wide range of animal hosts12–14 and in humans, causing wound and urinary tract infections, peritonitis, or endocarditis.15–18 M. sciuri and closely related species have been found to be an important reservoir of antimicrobial resistance in animals including livestock, 19 dogs, 20 and dromedary camels.21,22 Indeed, M. sciuri shares ecological niches, including its host nasal cavity with closely related multidrug-resistant Staphylococcaceae such as S. aureus among others, which is a favorable environment for the transfer of genetic elements. Such elements can be mobilized and transmitted to other microorganisms via HGT events 15 or transduced by phages. 6
Despite its relevance, the prophage content of M. sciuri remains largely unexplored, limiting our understanding of its role in horizontal gene transfer and phage-mediated evolution. To address this gap, we investigated the M. sciuri prophage content in 26 strains. We built on previous studies23,24 describing 26 complete M. sciuri genomes derived from isolates from Kenyan camels and dogs to investigate the diversity of temperate M. sciuri phages from Africa. We successfully isolated eight novel temperate phages and characterized them both morphologically and genetically using transmission electron microscopy (TEM) and whole genome sequencing methods. In total, the eight phages were classified into two new families, comprising three novel genera. Phages Φ6228, Φ6223, Φ6229, and Φ031 were grouped in one family, while Φ6255, Φ036, Φ116, and Φ127 belong to the other.
Materials and Methods
Bacterial strains and culture conditions
Ten strains of M. sciuri isolated from camels 24 and 16 strains of canine origin 23 were used in this study (Table 1). All strains were plated on Trypticase soy agar (BD, Switzerland) and incubated overnight at 37°C. Single colonies were then picked and grown in Trypticase soy broth (BD, Switzerland) media at 37°C while shaking at 220 RPM. Phages were amplified using the double-layer-overlay method, as described previously. 25
Kenyan Mammaliicoccus Sciuri Strains Used in This Study
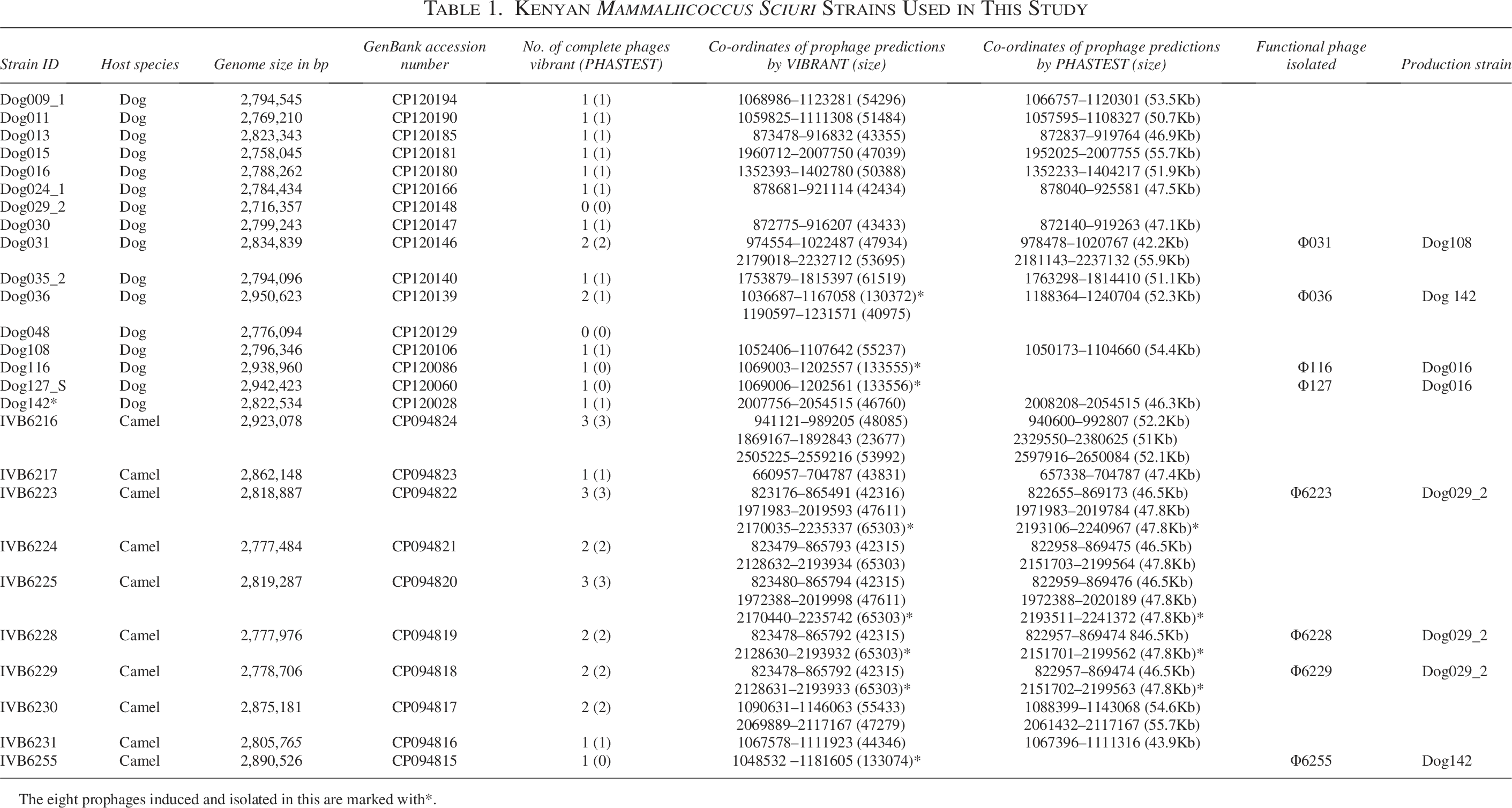
The eight prophages induced and isolated in this are marked with*.
In silico detection of temperate phages in M. sciuri genomes
All 26 M. sciuri genomes were analyzed using the online tool PHASTEST 26 and the software VIBRANT v1.2.1. 27 The outputs of these two tools in terms of the number of prophages detected and their sizes were compared, and different individual prophage sequences were used for subsequent analysis.
Predicted prophage sequences were annotated using Pharokka 28 v1.7.0 and phylogenetic trees were constructed using the software VICTOR (Virus Classification and Tree Building Online Resource) 29 applying the trimming method and the D0 formula.
Phylogenetic analysis of the eight isolated temperate phages
The dataset for protein clustering and phylogenetic analysis was initially identified from vConTACT3 using the INPHARED January 1, 2025 database as the input dataset. 30 These data clustered the isolated M. scuri phage genomes into two distinct sets. The first consisted of a total of 137 genomes and the second four genomes. Prior to further analysis, all genomes were screened with CheckV, 31 and sequences identified as fragments or containing ≥10% contamination were excluded. Three genomes (PQ281812, OQ578994, and OR479082) were subsequently excluded based on these results. A hierarchically clustered tBLASTx distance tree of these 137 genomes alongside all existing Caudoviricetes genomes classified by the International Committee on Taxonomy of Viruses (ICTV), 6,672 genomes in total, was created using ViPTreeGen version 1.1.3. 32 In parallel, the 137 genomes were re-annotated using Pharokka v1.7.0 with Prodigal as the gene prediction tool28,33 to create a standardized annotation dataset. Protein sequences were clustered with MMSeqs2 34 using parameters of 50% coverage and 50% sequence identity to create protein clusters. A binary matrix was produced to indicate the presence or absence of each protein cluster by genome and was used to calculate pairwise Jaccard similarity values via custom Python scripts. Signature proteins were defined as those protein clusters present in all genomes and were aligned with MAFFT 35 and used to create partitioned maximum likelihood phylogenetic trees. Tree calculation was performed with IQTree2 36 using ModelFinder, 37 1000 ultra-fast bootstrap 38 and SH-aLRT test replicates. All trees were visualized and annotated using iTOL. 39 To assess relationships between these phages at the nucleotide level, inter-genomic similarity values were calculated using taxmyPHAGE. 40
Phage induction, isolation, and determination of phage infectivity
Prophages were induced using mitomycin C as previously described. 41 Briefly, mitomycin C was added to a final concentration of 1 μg/mL to bacterial liquid cultures in early exponential growth phase (OD600 ≈ 0.3–0.5). The liquid cultures were incubated for 8 h at 37°C and 225 rpm, then centrifuged at 10,000 × g for 10 min, and supernatants were passed through a 0.22 μm pore size filter. Filtered supernatant (5 μL) was spotted on double-layer agar containing the test strains. When plaques were observed, phages were picked from plaques using a pipette tip, resuspended in 200 µL saline magnesium buffer pH 7.5 (100 mM NaCl, 8 mM MgSO4·7H2O, Tris-Cl 1 M pH 7.5), and amplified using the double-layer-overlay method. 25 To investigate phage clonality, phage sequences were compared on the nucleotide level using BLASTn from the NCBI, applying standard settings. 42 Partial prophage induction was not investigated.
To determine the host range of each isolated phage, an efficiency of plating (EOP) assay was performed as previously described.
43
Phage lysate containing 108 plaque-forming units (pfu) was prepared for each phage, and serial dilutions were prepared in a 96-well plate. Five microliters of each phage dilution was dropped and allowed to dry on the solidified soft agar. Plates were incubated overnight at 37°C, and plaques were counted to determine phage infectivity. The software GraphPad Prism v.10.2.3 was used to display the results. Phage efficiency was adjusted to the highest plaque number on a given bacterial strain for each phage tested and was displayed using the following formula:
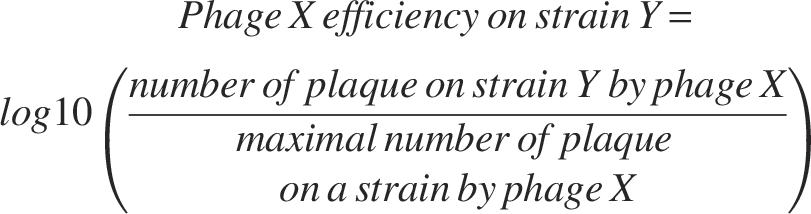
Concentration and purification of phages by cesium chloride gradient centrifugation
For TEM imaging, phage particles were concentrated by precipitation using polyethylene glycol (PEG8000) and purified by cesium chloride (CsCl) gradient centrifugation based on previously published protocols.44,45 Briefly, 6 mL of phage lysates harboring 109–1011 pfu/mL were overlaid onto three layers of CsCl gradient, with densities of 1.7, 1.5, and 1.3 g/mL, respectively. Samples were centrifuged in ultra-clear centrifuge tubes (14 × 89 mm, Beckman & Coulter, Inc., USA) in an SW41 Ti rotor (Beckman & Coulter) using a HITACHI CP100NX ultracentrifuge at 100,000 × g at 4°C for 2 h.
Phage DNA extraction, MinION, and Illumina sequencing
Phage DNA was extracted following the protocol “Fast & efficient isolation of phage genomic DNA using the Monarch HMW DNA Extraction Kits” (NEB). 46 Briefly, phage lysates containing between 109 and 1011 pfu/mL were treated with a final concentration of 5 µg/mL RNase A and 10 µg/mL DNase I for 60 min at 37°C. Next, the lysates were treated with 300 µL HMW gDNA tissue lysis buffer and 20 µL proteinase K for 45 min at 56°C. Proteins were then precipitated by adding 300 µL protein separation solution and centrifuged at 16,000 × g for 10 min. Finally, DNA was precipitated from the supernatant by adding 550 µL isopropanol, and DNA pellets were washed twice with 70% ethanol solution.
Two complementary approaches were employed for next-generation sequencing (NGS): Illumina MiSeq sequencing (v2, 300 cycles) using the Illumina DNA prep kit and Oxford Nanopore Technologies sequencing using the ligation prep kit (SQK-NBD114.24), MinION Flow Cell (R10.4.1), on a GridION X5 device. In each case, genomic DNA samples were multiplexed together according to manufacturers’ protocols for library preparation and sequenced on the respective platforms. For Nanopore data, demultiplexing and basecalling were done while sequencing live using the GridION X5 device under high-accuracy (HAC 400 bps; dorado version 7.2.13) mode, and sequencing was stopped after six hours when most samples reached at least 10,000 reads. Both Illumina MiSeq and GridION X5 sequencing were carried out at the Institute for Infectious Diseases, Bern, Switzerland.
Nanopore-based adaptive sampling was implemented to overcome the challenge of limited starting material. For this end, lambda DNA was introduced as a placeholder to increase sequencing depth, and the surrogate DNA was subsequently removed using adaptive sampling techniques akin to Nanopore Adaptive Sampling with Carrier DNA. 47 All sequencing was done using barcodes and the phages.
Hybrid assembly and phage genome annotation
An initial filtering step using SeqKit v1.10 48 was used to remove Nanopore reads below 1 kilobase in length, ensuring a focus on longer sequence data for improved assembly accuracy. The Flye assembler v2.9, 49 operating in metagenomic mode, was employed for unbiased de novo assembly of the phage genomes (flye –threads 20 –nano-hq –meta). 50 Post-assembly processing involved cleaning and validation steps using Phables v1.4.0 51 to refine the assemblies.
Polishing was conducted with short-read data from the MiSeq platform, utilizing Polypolish v0.6.0 52 to improve sequence accuracy. The assembled viral sequences and overall quality were assessed using CheckV v1.0.3. 31 Phage genomes were annotated using Pharokka 28 v1.7.0 and visualized using Clinker. 53 All tools were used with basic settings unless specified otherwise.
Transmission electron microscopy
A 10 µL volume of high-titer phage lysate (109–1011 pfu/mL), resuspended in 200 mM sodium chloride, 10 mM MgSO4, 50 mM Tris, and 0.01% gelatine at pH 7.4, was placed on glow-discharged and Formvar-coated grids (Ted Pella Redding, Redding, California, USA) in 0.25% Dichloroethane (Merck, Dietikon, Switzerland). After 2 min incubation, excess liquid was removed from the grid with filter paper, and the phages were washed twice with 0.1 M sodium cacodylate solution. The grid was then incubated with 10 µL Uranyless (Delta Microscopies, Mauressac, France) for 30 s, and, after a drying period of at least 2 h, the grid was transferred to a Talos 120C transmission electron microscope (Thermo Fisher Scientific, Hillsboro, Oregon, USA) at 120 kV. Images were collected at magnification of 73000×.
Results
In silico screening and phage induction of East African M. sciuri reveals the presence of intact prophage sequences
To unveil the diversity of prophage sequences of nonclinical Staphylococcaceae, we screened the genomes of 26 East African M. sciuri strains previously isolated from camels and dogs. Complete prophage sequences were found in 24 strains (Table 1), while two strains (Dog29_2 and Dog048) had no prophage detected. On average, 1.38 prophages were found per M. sciuri genome, while the number of prophages found in M. sciuri strains from camels was higher than those from dogs (2 prophages detected per genome versus 1). The results obtained by the two software tools were 89% identical. PHASTEST detected 32 prophage genomes in 26 strains, while VIBRANT predicted 36 prophage regions in 26 strains (Table 1). We classified these prophages phylogenetically, and their sequences clustered in seven groups and one singleton (Fig. 1). Interestingly, all phylogroups had prophages originating from strains isolated from both camels and dogs, except for phylogroups G3 and G6 that were only found in camel-derived strains. PHASTEST was not able to detect all four prophages belonging to phylogroup G7.
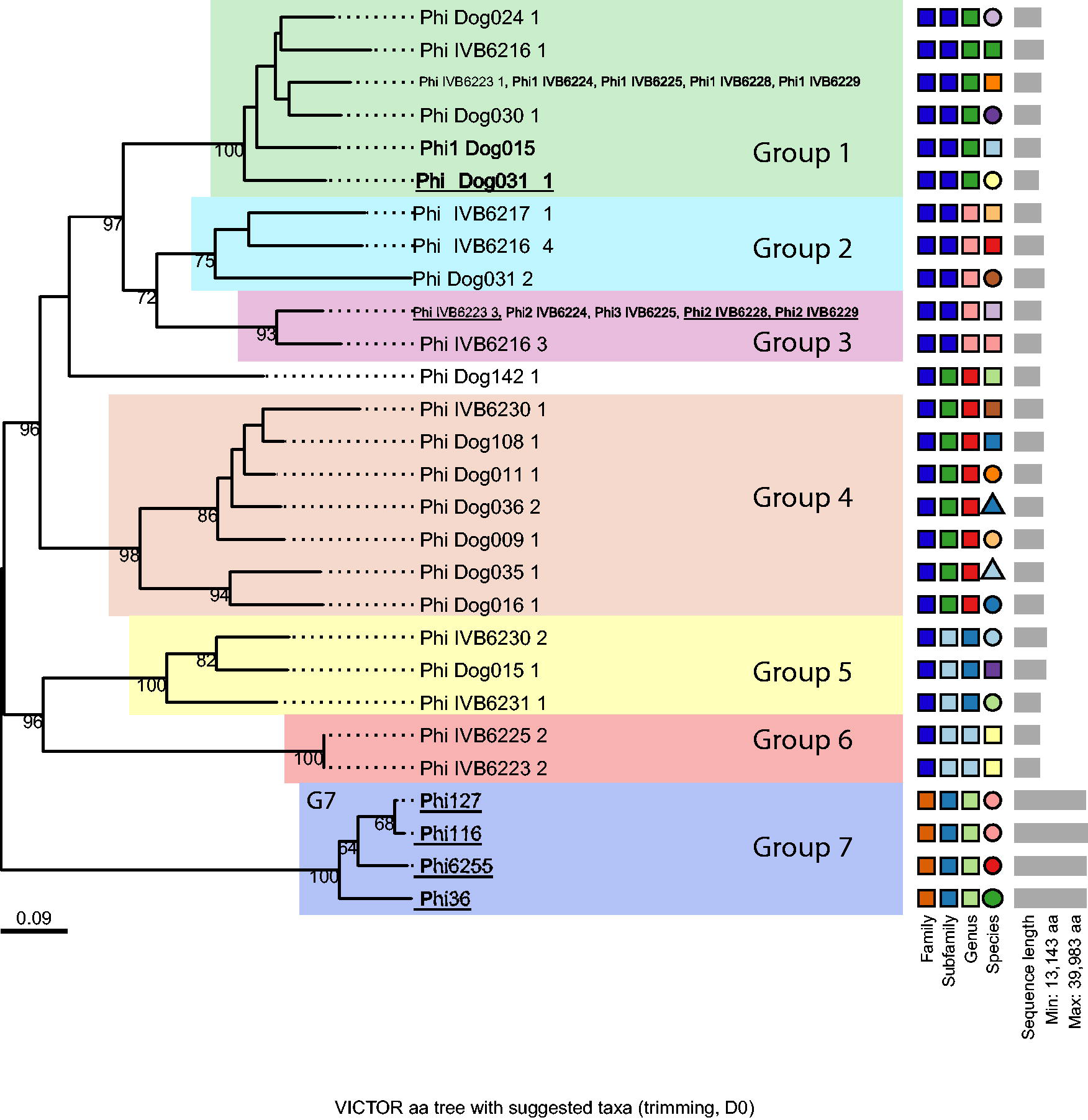
Phylogenetic tree of 36 complete prophage sequences identified in silico in 26 Kenyan M. sciuri strains. Prophage sequences were classified using VICTOR with the D0 formula. They are clustered into seven groups highlighted by colored boxes. Bootstrap values from 100 iterations are displayed. The phages successfully induced and isolated are underlined. Each prophage is named as follows: Phi_Name of the strain_number of the prophage. VICTOR, Virus Classification and Tree Building Online Resource.
Induction of eight functional phages from three different groups: G1, G3, and G7
The presence of putative intact prophage sequences in almost all M. sciuri genomes led us to assess their potential to be induced using mitomycin C. Eight phages out of the 36 putative intact prophages predicted in silico were successfully isolated after induction and subsequently characterized. Each phage was recovered from a different strain. All recovered phages belonged to the phylogroups G1, G3, and G7 (Fig. 1, Table 2). Besides those belonging to the phylogroup G7 (n = 4), predicted as two incomplete phages with scores <65, all the other recovered phages (n = 4) had scores of 150 given by PHASTEST. PHASTEST was more precise in predicting the prophage size. As an example, VIBRANT predicted the genome size of phages Φ6223, Φ6228, and Φ6229 to be 65 kbp, while PHASTEST predicted a size of 48 kbp for those phages, which is close to the experimentally verified phage genome sizes of 44, 47, and 44 kbp for phages Φ6223, Φ6228, and Φ6229, respectively. With respect to phage Φ31, PHASTEST gave a predicted size of 42.2 kbp (which is nearly the same size as experimentally verified phage genome size), while VIBRANT predicted a prophage genome size of around 48 kbp. Overall, both tools revealed comparable results (Table 1). The employed phage induction protocols did not necessarily result in induction of genetically similar prophages. This was particularly striking for the phylogenetic group G3 where phages Φ6223, Φ6228, and Φ6229 could be recovered while the other two could not (Fig. 1). Similarly, phage Φ31 was the only recovered phage from phylogenetic group G1, which consisted of ten predicted phages (Fig. 1). In contrast, all phages belonging to the phylogenetic group G7 were successfully isolated after the induction experiment.
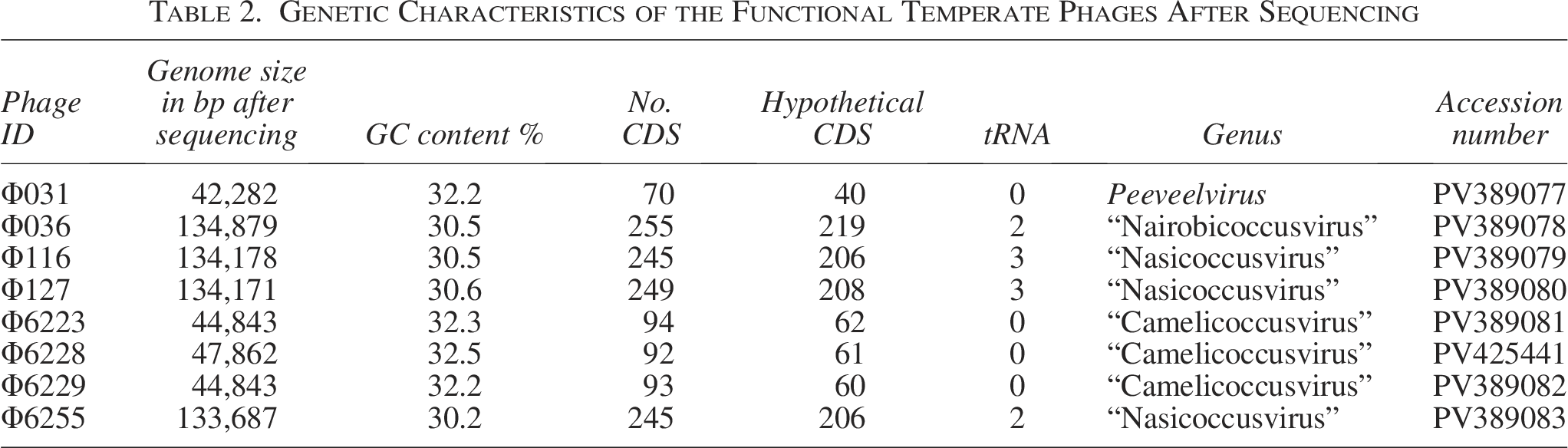
Genetic Characteristics of the Functional Temperate Phages After Sequencing
All induced phages have a siphovirus morphology
TEM images were obtained for the eight isolated phages, and typical representatives are displayed in Figure 2. All phages shared a classical siphovirus structure according to the Ackermann classification. 54 However, small differences were observed in the morphology of the capsid shape, the length of their respective tails, their tail fibers, and tail spikes (Fig. 2 and Supplementary Table S2). Phages Φ31, Φ36, Φ116, Φ127, and Φ6255 were characterized by a typical icosahedral head, while the other three (Φ6223, Φ6228, and Φ6229) possessed prolate heads (Fig. 2). Phage measurements (tail length, head length, and head width) as well as statistical tests to show size differences are reported in Supplementary Table S2.

Representative transmission electron micrographs of three phylogenetically different phages induced and characterized in this work. Magnification of each micrograph is 73,000×.
Phylogenetic analysis shows phages Φ36, Φ116, Φ127, and Φ6255 belong to a new family, while the four others belong to a second new family
The bipartite network produced by vConTACT3 indicated that the induced M. scuri phages formed two distinct groups, referred to here as clades 1 and 2. Clade 1 comprised Φ6228 (PV425441), Φ6223 (PV389081), Φ6229 (PV389082), and Φ031 (PV389077) and included a further 133 genomes. Examination of the tBLASTx distance tree (Fig. 3) and protein clustering results (Supplementary Fig. S2) indicated that when using protein clustering thresholds of 50% sequence identity and coverage, the 137 genomes could be divided into six deep-branching clades. We propose that each of these clades represent new families within the class Caudoviricetes. The M. scuri phages Φ6228 (PV425441), Φ6223 (PV389081), Φ6229 (PV389082), and Φ031 (PV389077) formed one of these clades that also included Staphylococcus phages phi575 (KY389063) and phi879 (KY389064) (Fig. 4A). Eight core proteins were identified amongst these phages, with the ML phylogeny reflecting the structure of the tBLASTx distance tree. According to the recommended ICTV demarcation criteria, 55 Φ6228 (PV425441), Φ6223 (PV389081) and Φ6229 (PV389082) form a single species within a new genus, while Φ031 (PV389077) represents a new species in the genus Peeveelvirus. We proposed “Mammaliicoccusviridae” and “Camelicoccusvirus” as new family and genus names, respectively.
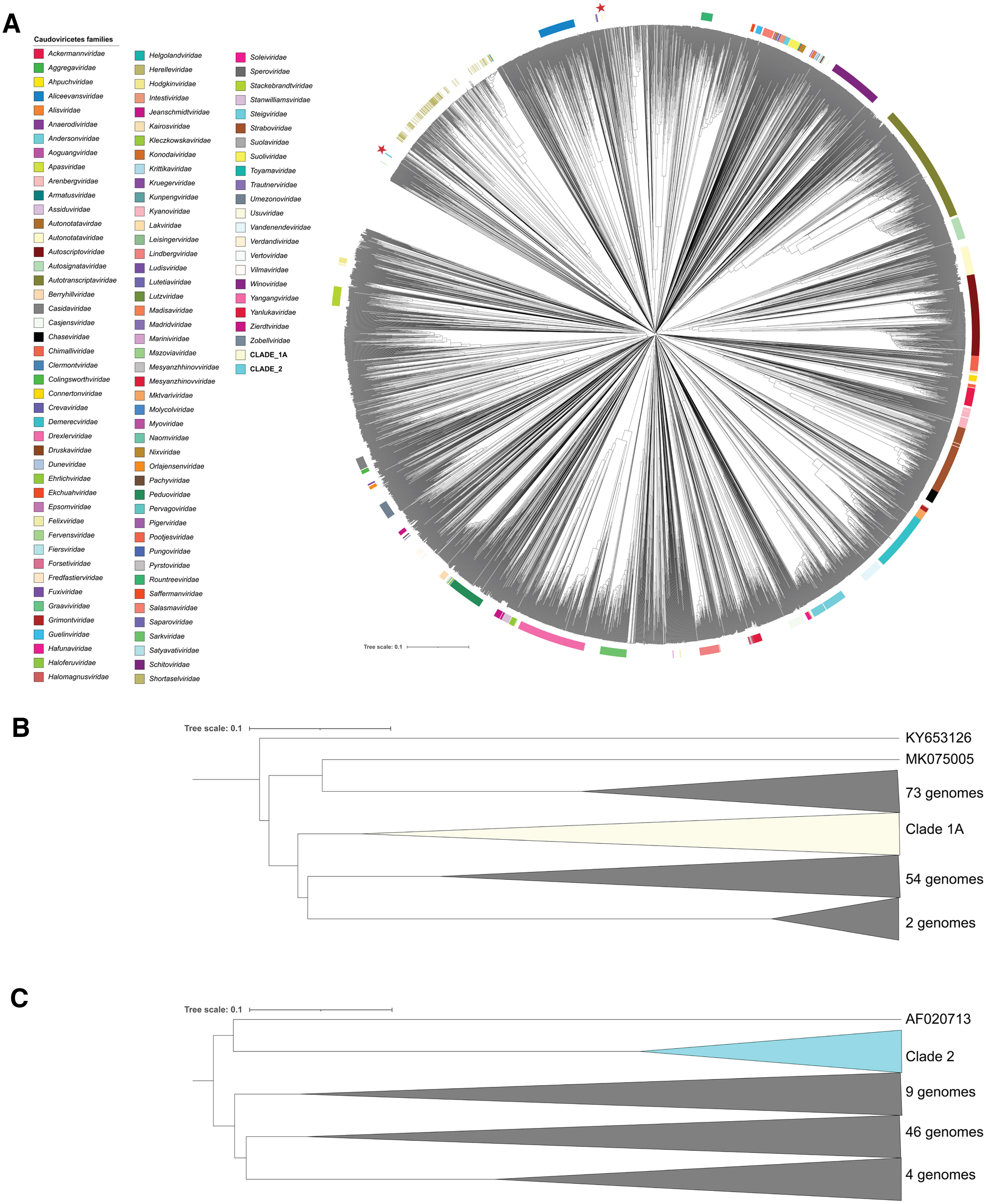
tBlastx distance tree of the 8 functional temperate M. sciuri phages and all phage from the ICTV Virus MetaData Resource.
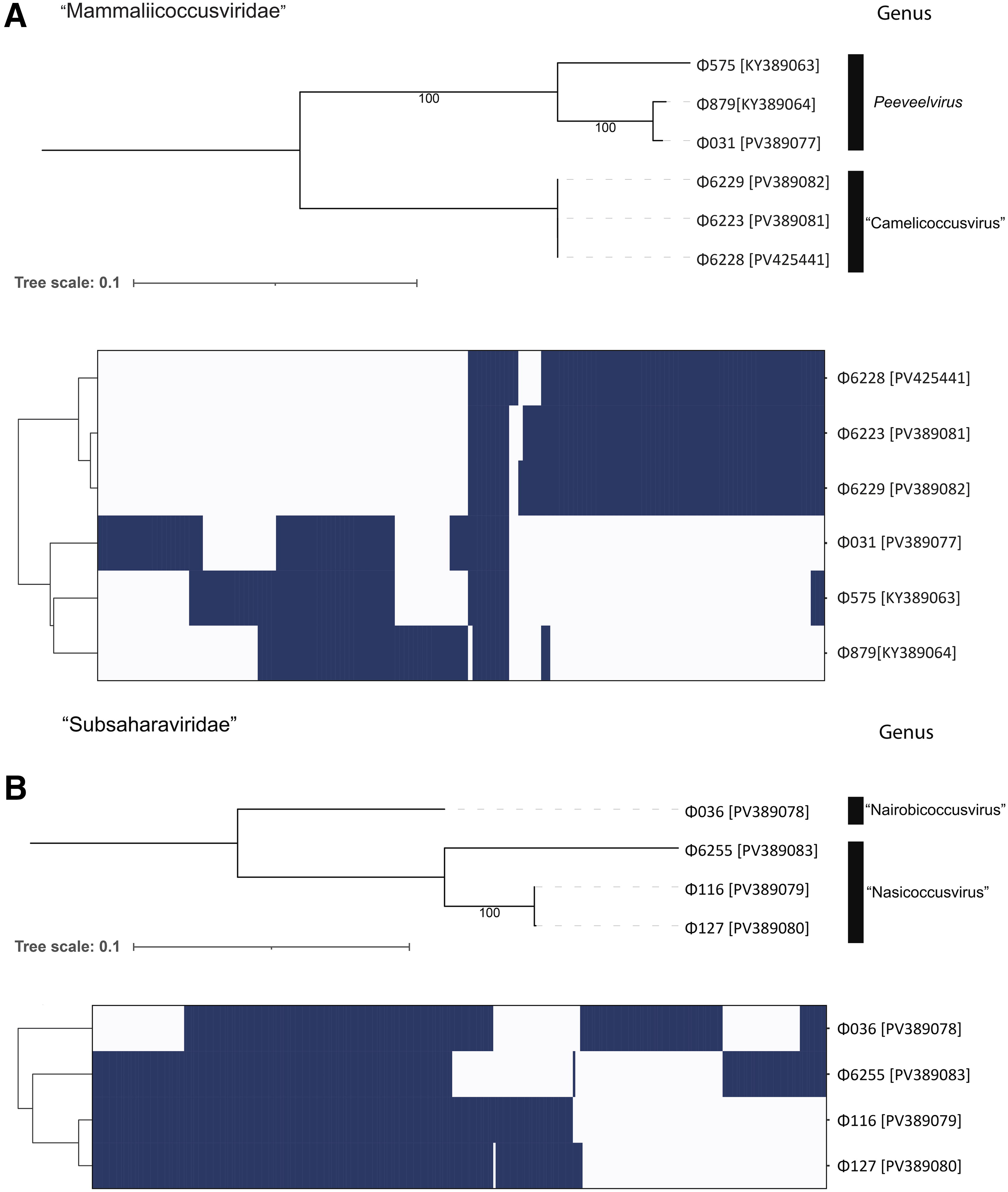
Phylogeny and protein clustering of the proposed families “Mammaliicoccusviridae”
The second clade consisted of Φ6255 (PV389083), Φ036 (PV389078), Φ116 (PV389079), and Φ127 (PV389080). A total of 111 core protein clusters were identified, representing an average of 56% of protein coding sequences for each phage (Fig. 4B). These phages form two genera consisting of one and two species, respectively. We proposed “Subsaharaviridae” as the name for this family, which includes two new genera, “Nairobicoccusvirus” and “Nasicoccusvirus.”
Phages Φ6223, Φ6228, and Φ6229 have genome sizes that range from 44 to 48 kbp and were all inserted in the same location of the M. sciuri genome, downstream of a MarR family transcriptional regulator and an
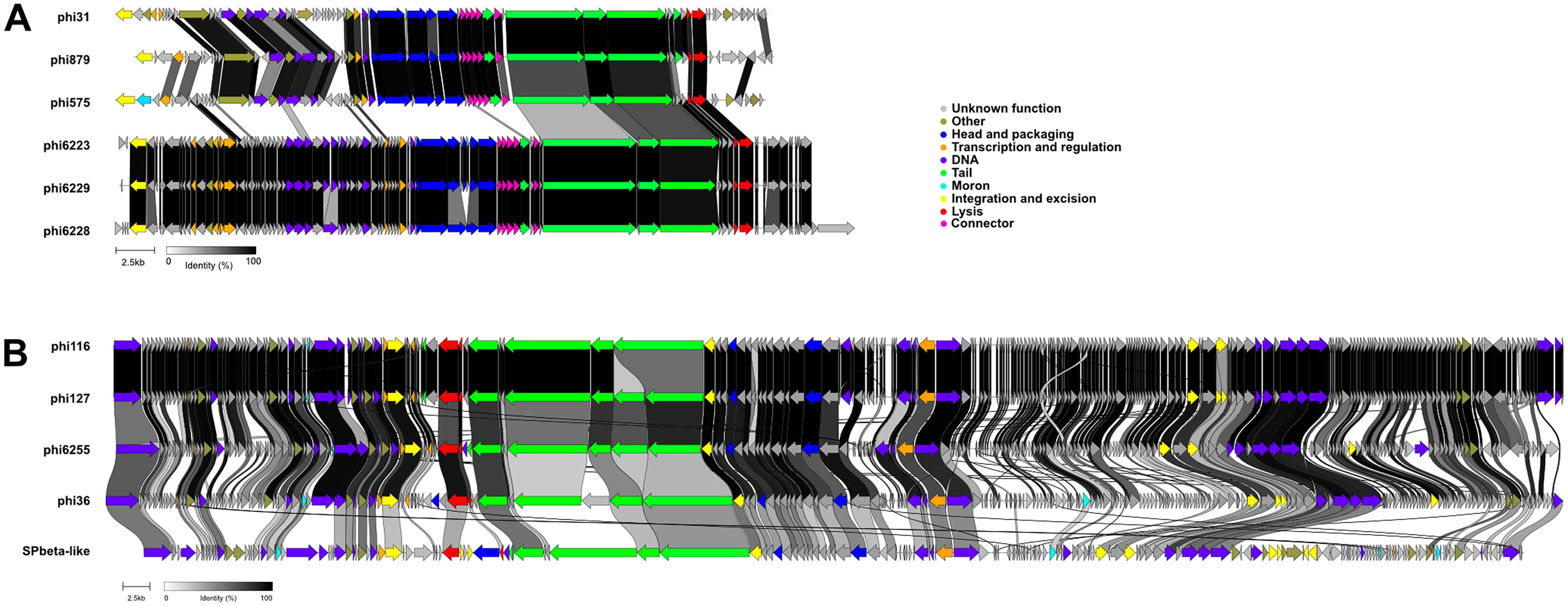
Genome comparison of phages belonging to the new proposed families. Alignments of phage genomic sequences from new proposed family “Mammaliicoccusviridae”
Phages Φ36, Φ116, Φ127, and Φ6255 have large genomes ranging from 133 to 135 kbp in size (Fig. 5B). The phages Φ116 and Φ127 were nearly identical to each other, as both prophages had high similarity sequences originally in their respective host strains. Φ6255 had about 80% similarity at the DNA level to phages Φ116 and Φ127, while Φ36 had only 70% similarity to phages Φ116 and Φ127. A BLASTn analysis against all other known phage genomes deposited in GenBank database confirmed their relatedness with the S. epidermidis phage named SPβ-like 56 (KT429160.1). For all phages of this group, between 84% and 85% of the genes encode proteins of unknown function. However, their overall phage genome organizations were very similar (Fig. 5B).
Phages from clade 2 had the same insertion site in M. sciuri genome. They were inserted at the very end of a gene coding for a bifunctional GNAT family N-acetyltransferase/carbon-nitrogen hydrolase family protein.
Phages from clade 2 show a broader host range but produce generally lower titers
Finally, we thoroughly assessed productive infection of all eight isolated phages against the different 26 African M. sciuri strains as defined by EOP (Fig. 6). We were able to identify two main patterns regarding the host range of the M. sciuri phages isolated in this work. Phages belonging to the first phylogenetic clade 1, namely Φ6223, Φ6228, and Φ6229, had a relatively narrow host range with a capacity to infect five to six M. sciuri strains. However, they were able to produce high titers on the susceptible strain Dog29_2. Phage Φ31 had a broader host range, with 17 strains infected and the highest plaque number. Phages belonging to the clade 2 (Φ36, Φ116, Φ127, and Φ6255) had a broad host range while remaining moderately infectious on all the susceptible strains. With 18 M. sciuri strains susceptible, the closely related phages Φ116 and Φ127 were the most infectious, while Φ36 successfully infected 10 M. sciuri strains.
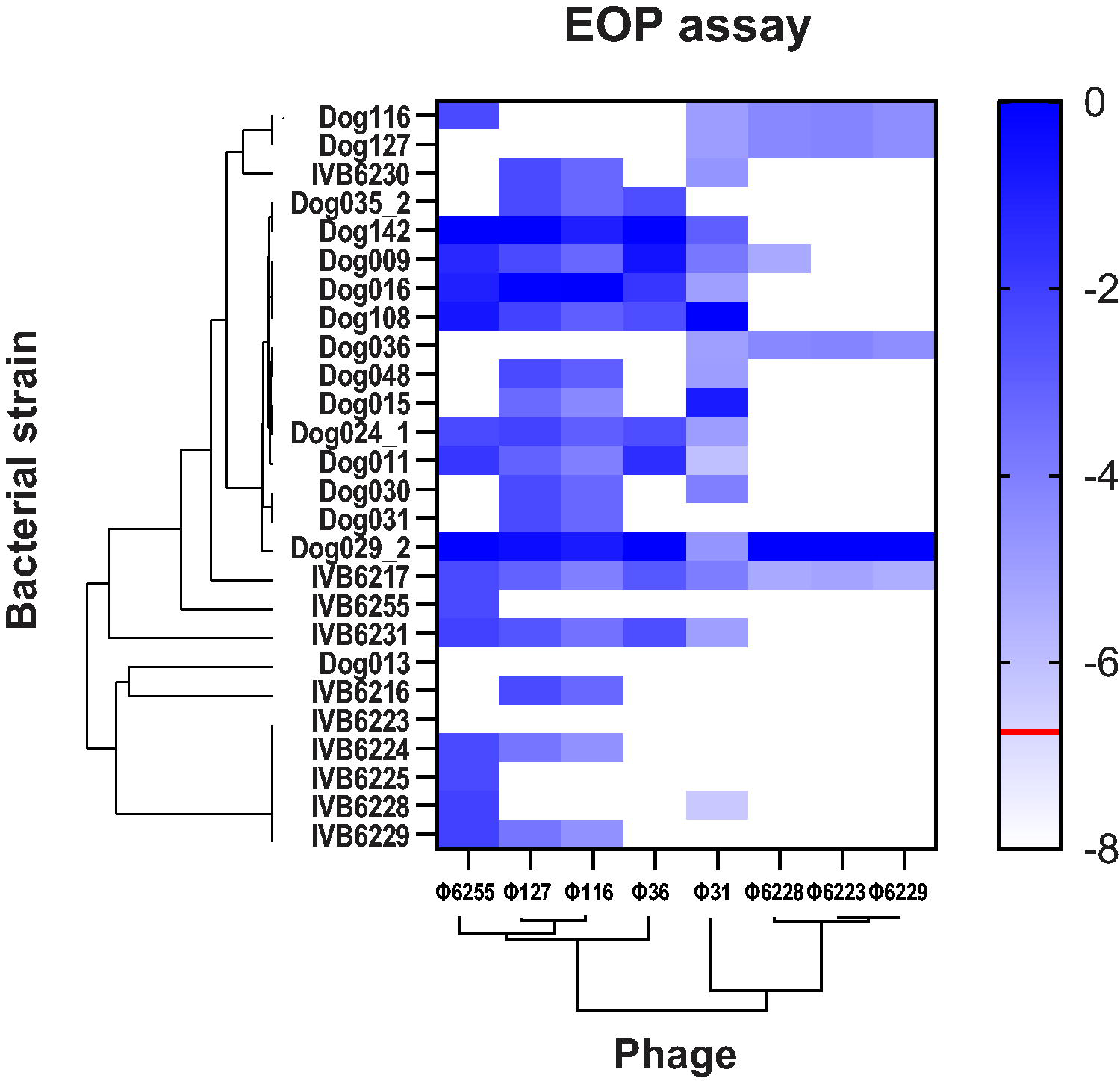
Determination of the host range of all eight phages on different M. sciuri strains. EOP assay for the eight characterized M. sciuri phages was performed on each M. sciuri strain. Reference value (0) was set to the most susceptible bacterial strain of each phage. The value −1 indicates 10 times less plaque than the highest number of plaques shown by a phage on a bacterial strain. The phylogenetic tree of the phages was created using VICTOR online research tool while the bacterial phylogenetic tree was based on MLST sequences. The red line shows the limit of detection. White rectangles indicate that no plaques were detected. EOP, efficiency of plating.
Significant differences were also observed regarding the susceptibility of the M. sciuri strains to phage infection. Strains IVB6216, IVB6224, IVB6225, IVB6228, IVB6229, IVB6223, and Dog013 (Fig. 6) were resistant to numerous phages with the latter two being resistant to all phages isolated in this study. In contrast, M. sciuri strains Dog035_2 and IVB6217 were susceptible to all phages tested in this work.
Discussion
This work aimed to increase our knowledge of the prophage diversity and dynamics within the genomes of M. sciuri strains isolated from Kenyan dogs and camels. First, our in-silico analyses revealed the presence of 36 prophage genomes in 24 M. sciuri strains, which is in line with similar previous studies in S. aureus. 57 Only two Kenyan strains had no apparent prophages in their genomes, regardless of which predictive software was used (Dog29_2 and Dog048). Induction experiments showed no change in OD600 after addition of Mitomycin C compared to controls for these strains. The absence of lysis on all bacterial strains tested following spot tests with supernatants derived from those two strains also supported the absence of complete inducible prophages. M. sciuri strain Dog029_2 was particularly sensitive to phage infection and was used to amplify three temperate phages investigated in this study. This is not surprising, as bacterial resistance to phage infection is generally correlated with their prophage content due to superinfection exclusion mechanisms. 58 In total, eight new viable siphovirus prophages belonging to two new taxonomic families were isolated. Their identification and characterization expand the current phage catalogue for this species, which despite its potential clinical relevance, few lytic or lysogenic phages have been reported in the literature. 6
Phylogenetic analyses revealed that the phage Φ31 was closely related to the previously identified M. sciuri phages Φ879 and Φ575, known for their capacity to transduce plasmids encoding staphylokinase, phospholipase, and SCCmec elements among others 6 (Fig. 4A). This might suggest that Φ31 may have a similar capacity with respect to HGT within M. sciuri populations, further facilitated by its broad host-range within the species, as it was active on ∼65% of the M. sciuri strains used in this work. Further experiments will be required to assess the HGT potential of this phage in M. sciuri and other related species.
The closest phylogenetic relative to phages from clade 2 is a 127-kbp temperate phage named SPβ-like isolated from S. epidermidis (GenBank KT429160.1). SPβ phages were originally isolated from Bacillus subtilis strain 168. 59 They are characterized by large genomes carrying multiple integrases and regulatory elements, which hints towards a complex interplay between lytic and lysogenic cycles that may enhance their ability to persist and propagate within their bacterial hosts. 60 Other phages with large genomes were previously identified in Staphylococcaceae species, such as those that belong to the well-characterized genera Kayvirus 9 and Silviavirus. 8 The lytic phages belonging to these genera have been characterized by their broad host ranges within the species S. aureus including lytic activities against biofilm-producing and methicillin-resistant isolates.61,62 Phages Φ116, Φ127, and Φ6255 from this study seem to have a broad host range since they were able to infect between 60 and 70% of the 26 M. sciuri strains investigated in this study. Despite its phylogenetic relatedness with the three “Sciuriphagovirus” phages, Φ36 has a narrower host range and is only active against 10 M. sciuri strains (38%). It would be interesting to further characterize the genetic elements of the phages presented here and previously characterized phages to improve our understanding of the phage–host interactions among Staphylococcaceae species. Advances in synthetic genomics now enable the genetic modification of phages, 63 such as the successful removal of the lysogeny control region of a Listeria phage, converting it into an obligatorily lytic phage. 64 Systematic site-directed modifications of phage genes followed by subsequent phenotypical testing will allow us to get more mechanistic insight into the phage physiology including novel phages like the ones reported here. The continuing search for new temperate phages will deepen our understanding of phage diversity and dynamics. Finally, novel phages with a broad host spectrum might provide a blueprint for genetically engineered therapeutic phages in the future.
Authors’ Contributions
J.D.R.C. wrote the original draft of the article and performed all experiments related to phage inductions, isolations and purifications as well as host range determinations with assistance from T.M.W. and S.T.-P. J.L. and S.K. did the TEM imaging. L.B. and A.R. did the sequencing and assemblies of the phage genomes. D.T. and P.K. and performed the phylogenetic analyses, and J.E.F. did the bioinformatic analysis from the Supplementary Data. J.J. and F.L. designed the study and acquired the funding for this work. All the authors reviewed, edited and approved the final article.
Footnotes
Acknowledgment
The authors thank Laurent Fischer for their valuable assistance in formulating the new family- and genera names.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Funding Information
This work was funded by the Multidisciplinary Center for Infectious Diseases (MCID) of the University of Bern (Grant ID: MA-11). This work received additional support from the University of Bern and a research grant from the “Leading House South Asia and Iran, Zurich University of Applied Sciences. TEM imaging was performed at the Microscopy Imaging Center (MIC), University of Bern, Switzerland.
Data Availability Statement
All raw sequencing data can be found in Bioproject PRJNA1175884.
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.