
Abstract
Keywords
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fatal, progressive disease that causes degeneration of cortical, brainstem, and spinal cord motor neurons leading to both upper and lower motor neuron dysfunction.1,2 Among the symptoms are muscle weakness, spasticity, dysphagia, dysarthria, and respiratory failure. ALS is also associated with frontal temporal dementia (FTD) and more subtle cognitive impairment. The lifetime risk of developing ALS is approximately 0.27% with the annual incidence being 1-2 per 100 000 people. The lifetime risk of developing ALS is similar to multiple sclerosis (1 in 700 people).1,2
ALS has a genetic component with multiple genetic variants reported in previous work, but the cause for around 90% of the cases remains unknown even though age, veteran status, and various environmental factors have been shown to have an association with development of the disease. 2 Elevated pro-inflammatory markers such as interleukin 6 and Tumor Necrosis Factor-alpha have been reported in the sera of both mice and humans with ALS. 3 Research has shown neuroinflammation caused by T-lymphocytes and activated macrophages plays a key role in perpetuating motor neuron death in the disease. 4 However, the cause of neuronal loss and denervation remains unclear. A cure for the disease is yet to be discovered, therefore current treatment options focus on symptom management and supportive care. 5
Blood purification through plasma exchange (PE) is commonly used to treat a number of neurological conditions where removal of circulating autoantibodies, neurotoxic or pro-inflammatory factors play a major role. 6 Recently, a pilot clinical trial carried out on 13 patients in Spain has revisited PE as a potential therapy for ALS, 7 more than three decades after some early studies were performed.8–11 In the Spanish trial, human albumin was used as the replacement fluid because of its role in blood as a transporter of molecules 12 and its antioxidant, anti-inflammatory, and neuroprotective effects.13,14 Results indicated that about two-thirds of the patients studied showed a slower than expected rate of decline at the end of treatment. Whether those outcomes where attributable to a real beneficial effect of PE or to the heterogeneity of ALS evolution in the particular population analyzed remained unclear. To offer further insight, a similarly designed pilot study was carried out in the US.
Methods
Objectives
This was a phase 2, prospective, open label, single arm, non-controlled, single center, pilot study (ClinicalTrials.gov ID: NCT02872142) whose primary objective was to investigate the efficacy of therapeutic PE using albumin as the replacement fluid (PE-A) in slowing ALS progression over the course of a year using the revised ALS Functional Rating Scale (ALSFRS-R) and Functional Vital Capacity (FVC).
Safety and tolerability objectives of the PE-A procedure were assessed by monitoring adverse events (AEs), clinical laboratory testing, physical examination, and vital signs.
All procedures of the study were compliant with the standards for Good Clinical Practice (GCP), local laws and regulations, and the Declaration of Helsinki. The protocol and associated documents were reviewed and approved by the Committee for the Protection of Human Subjects (CPHS) at Dartmouth-Hitchcock Medical Center (reference number: STUDY00029255).
Patients
Patients of both sexes between 18 and 70 years old, with a possible, probable, or definite diagnosis of ALS according to the revised El Escorial criteria, 15 having experienced their first ALS symptom within 18 months prior to recruitment/consent, and with FVC >70% were eligible for participation in this study. Written informed consent to participate in the study was obtained from each patient at the screening visit.
Exclusion criteria included patients with pre-existing clinically significant lung disease not attributable to ALS; diagnosed with other neurodegenerative diseases or diseases associated with other motor neuron dysfunction; pregnant or breastfeeding women; a contraindication to undergo PE-A (eg, problematic peripheral vein access, history of anaphylaxis or severe systemic response to albumin, heart disease), abnormal coagulation parameters; renal dysfunction (creatinine concentration >2 mg/dL); prior behavioral disorders requiring pharmacological intervention with less than 3 months of stable treatment; or any condition that would complicate compliance with the study protocol.
For the post hoc control comparison, data from 18 ALS patients were obtained from the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) consortium database. 16 Control patients were clinically matched by mean age, baseline ALSFRS-R and FVC scores, as well as the time since onset of symptoms to study enrollment. Only patients from the placebo arms were included. The inclusion criteria were also applied to the control group.
Treatment Plan
The active treatment participants underwent weekly PE-A sessions using albumin 5% (Albutein, Grifols Biologicals LLC, Los Angeles CA, US) as the replacement solution. The first 3 weeks were an intensive phase where the patients had 2 PE-A sessions per week. This phase was followed by a 21-week maintenance phase where the patients had 1 PE-A session per week. The patients were then followed for an additional 24 weeks for a total study duration of 48 weeks. In addition to baseline (visit 1, week 0), efficacy and safety endpoints were evaluated at 5 visits (weeks 4, 12, 25, 36, and 48).
The planned volume to be exchanged at each PE-A was 100% to 110% of the plasma volume based on the sex, weight, and hematocrit of the patient.
PE-A was to be performed by peripheral venous access at a rate of 40 to 100 mL/min with a continuous flow Cobe Spectra or Optia blood cell separator (both from Terumo BCT, Inc., Lakewood CO, US) using anticoagulant citrate solution A (ACD-A) in a 1:10 ratio and a continuous infusion of calcium gluconate at 1 g/h. If an extraction flow rate >45 mL/min could not be achieved on 2 consecutive PE-A sessions, the patient was offered the possibility of installing a central venous access line.
Efficacy Assessment
ALSFRS-R scores (range 0 to 48, with higher scores indicating better function)
17
and FVC (%) were measured at baseline and tracked throughout the duration of the study (follow-up visits at week 4, 12, 25, 36, and 48) and used as a measure of disease progression. For comparison of expected evolution of untreated ALS, it was considered that overall ALSFRS-R scores decline at a rate of −1 point/month, according to the European Medicines Agency (EMA) guidelines for the treatment of ALS.
18
The expected decrease was calculated according to the EMA formula:

Safety Assessments
Safety variables assessed in this study were: percentage of PE-A sessions associated with at least one adverse event (AE) during or within 72 h after the completion of the product infusion; percentage of PE-A sessions temporarily associated with at least one AE, irrespective of causality, during or within 72 h after the completion of the product infusion; incidence of all AEs; vital signs and pulse oximetry recorded at each assessment visit, before, during, and after each PE-A session; and clinical laboratory testing (coagulation, blood count, biochemistry, and serology) at each specified visit.
Statistics
Since this was a pilot study, no formal calculation of sample size was performed. Similarly to the twin study performed in Spain, 7 a number of 10 patients was considered adequate for the evaluation of the objective of the study.
The evaluable population was defined as all patients who received at least one PE-A treatment and had at least one baseline determination and a measurement of a primary efficacy variable at a subsequent visit. The per-protocol (PP) population was defined as all evaluable patients who completed the treatment without major protocol deviations which could impact the primary efficacy assessment.
A t-test (if the assumption of normality was met, or otherwise a non-parametric Wilcoxon signed-rank test) was used to compare the difference between the ALSFRS-R scores at each of the follow-up visits and the baseline to the expected decrease. The significance level was set at 0.05.
Results
Population Characteristics
A total of 12 of 15 patients who provided informed consent passed screening and were enrolled in the study and in the evaluable population. All 12 patients (100%) completed at least 10 PE-A during the study. Six patients (50%) completed all 27 planned PE-A as specified in the protocol (2 patients discontinued due to an AE, 1 patient died, and 3 patients withdrew consent). A total of 10 patients were included in the per-protocol population (2 patients excluded due to lack of treatment compliance). Figure 1 summarizes the flow of patients across the study.

Flow of patients across the study.
The mean (SD) age was 60.0 ± 7.6 years. Seven patients were male (58.3%). All the patients (100%) were white or Caucasian. Weight was 75.8 ± 16.4 kg, and height was 169 ± 11 cm.
Seven patients (58.3%) were diagnosed with probable ALS and 5 patients (41.7%) were diagnosed with definite ALS. The median time from diagnosis to enrollment in the study was approximately 1 month (IQR: 0.6, 6.1 months). The median time since symptom onset to diagnosis was 10.5 months (IQR: 8.6, 15.4). Six patients started with a bulbar ALS onset and 6 with a spinal ALS onset (50.0% each). The most frequent body part first affected by ALS was the bulbar region in 6 patients (50.0%), followed by right and left upper extremities (3 patients [25.0%] each).
ALS Functional Rating Scale-Revised
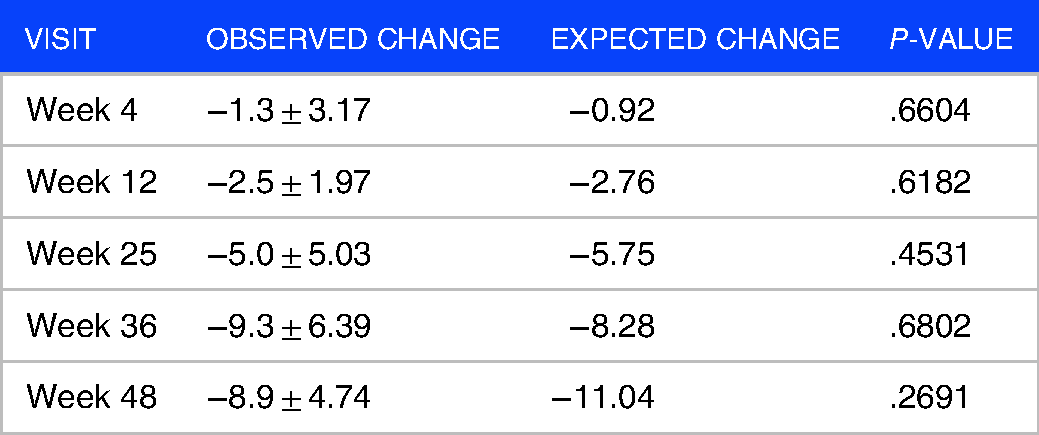
The median baseline value of the overall ALSFRS-R score was 43.0 (IQR: 35.3, 44.3). A progressive decline in the ALSFRS-R overall score was observed throughout the study in the evaluable population, consistent with a general increase of the functional impairment of patients during the study (Figure 2). Statistically significant decreases in overall score from baseline were observed starting in the treatment period at week 12 (mean [SD] change from baseline of −2.5 [1.97], P = .0020]) through the final follow-up visit at week 48 (mean [SD] change from baseline of −8.9 [4.74], P = .0026). There was no significant difference in ALSFRS-R scores between PE-A-treated patients and controls (Figure 2). Similar results were observed with the PP Population.

Distributions of ALSFRS-R scores at each of the 6 time points for patients and controls. N from week 0 to week 48 were for treated patients: 12, 12, 11, 7, 8, and 7. Dot: mean; Boxes: mean with Q1, Q3; Whiskers: minimum and maximum.
There were no statistically significant differences in the comparison of the change in ALSFRS-R score from baseline and that expected based on EMA guideline estimates (minus 1-point ALSFRS-R/month) for each of the follow-up visits (Table 1). However, according to the slopes of ALSFRS-R score decline, the “low progressor” patients were the most numerous (n = 6; 50%), followed by the “fast progressors (n = 4; 33%), and “normal progressors (n = 2; 17%).
Comparison of ALSFRS-R scores of PE-treated patients and EMA guideline estimates (mean ± SD).
Forced Vital Capacity
The median baseline FVC value was 87.5% (IQR: 45.3, 101.0). Analogously to ALSFRS-R, a progressive decline of pulmonary function was observed throughout the study (Figure 3). Statistically significant decreases from baseline in FVC (%) value were observed throughout the follow-up period at week 25 (−13.9 [7.01], P = .0020), at week 36 (−19.4 [12.44], P = .0031), and at week 48 (−21.0 [14.83], P = .0096]). Scores of control patients were like those of PE-A-treated patients (Figure 3). Similar results were observed with the PP Population.

Distributions of FVC scores at each of the 6 time points for patients and controls. N from week 0 to week 48 were for treated patients: 12, 12, 11, 7, 8, and 7. Dot: mean; Boxes: mean with Q1, Q3; Whiskers: minimum and maximum.
Safety
PE-A was performed using the peripheral route in 7 patients (58.3%) and primarily using central access in 5 patients (41.7%). Early discontinuation was reported in 2 and 3 patients from each route group, respectively.
Two patients (16.7%) had serious treatment emergent adverse event (TEAEs) (jugular vein occlusion and myocardial infarction). Three patients (25.0%) had AEs leading to treatment discontinuation (jugular vein occlusion, myocardial infarction, and venous thrombosis), and 2 patients had AEs leading to study withdrawal (jugular vein occlusion and venous thrombosis). One patient (8.3%) died of a serious, severe, non-TEAE of progression of ALS. All 12 patients had at least 1 TEAE during the study with the most common TEAEs (occurring in ≥3 patients [25%]) being dizziness (41.7%), fall (33.3%), and muscle spasms (33.3%). Most AEs were mild or moderate in severity with only 2 patients (16.7%) experiencing severe AEs (jugular vein occlusion and myocardial infarction).
A total of 59 of the 276 PE-A procedures performed (21.4%) were associated with TEAEs with 41 PE-A procedures (14.9%) associated with TEAEs considered related to study procedure and 22 PE-A procedures (8.0%) associated with TEAEs considered related to study drug and study procedure. None of the PE-A procedures were associated with study drug only.
Clinical laboratory, vital signs, and physical examination findings were unremarkable and consistent with changes expected because of the PE-A procedure or the patient's ALS.
Discussion
Similarly to the study performed in Spain, 7 the present study aimed to evaluate disease progression in patients affected by ALS and treated with PE-A with 5% albumin, without the limitations of earlier studies.8–11 We observed a progressive deterioration in both the co-primary endpoints ALSFRS-R scores and FVC over time, which indicate that the expected rate of decline due to ALS did not appear to have been slowed down by PE-A therapy. Similar results of limited efficacy of PE for the treatment of ALS were observed in all the previous studies.7–11 However, the good safety and tolerability of the PE procedure was confirmed in this fragile patient population.
This US study enrolled an older population in comparison to the study of Spain, 7 (means: 49 vs 60 years, respectively) and a lower proportion of patients with bulbar to spinal onset of disease (38%-62% vs 50%-50%, respectively). Bulbar ALS is the most devastating variant of the disease. 19 By contrast, time since symptom onset as a measure for disease aggressiveness 20 was shorter (medians: 13.6 vs 10.5 months). Despite such differential clinical profile of the patients in both studies, baseline ALSFRS-R and FVC values were similar (medians: 42 vs 43 points, and 87.5% vs 87.0%, respectively).
The progressive decline in the ALSFRS-R overall score observed throughout the study indicated a general increase in patients’ functional impairment. Statistically significant decreases in overall score were observed starting in the treatment period at week 12 through the final follow-up visit at week 48. Although the mean decreases from baseline at week 25 and week 48 were less than expected from ALS natural progression in untreated patients according to EMA guidelines, 21 these differences were not statistically significant. Similar results were observed in the study of Spain. 7 Regarding the coprimary secondary endpoint, a progressive decline in FVC was observed starting at week 12 and continuing through the follow-up period. Again, similar results were observed in the study of Spain. 7
In principle, a PE-based therapeutic approach to ALS may appear sound. Research on the neuroimmunology of ALS has shown increased inflammatory cytokine levels that lead to increased activation of pro-inflammatory monocytes which then perpetuate the cycle of neuron cytotoxicity. 22 Other studies showed that immunosuppressive regulatory T cells (Tregs) are also inhibited in ALS therefore leading to further neuroinflammation. 23 While PE to relieve Treg inhibition or remove the pro-inflammatory factors in patient sera could be a possible way to slow the decline, this study did not show any benefit, in line with the preceding pilot studies. However, an analysis of the patients’ cytokine profile may provide more concrete answers regarding the changes, or lack thereof, that PE may have caused to neuro-inflammatory markers. More targeted therapies or infusion of anti-inflammatory factors may prove more successful in slowing disease progression by reducing the pro-inflammatory milieu.
Because ALS is a heterogeneous syndrome, it is plausible that PE may be beneficial for only a subset of patients.
24
Given the multiple aberrant processes that lead to ALS that have been reported, some patients may respond to PE while others may not. Our results showing an unbalance towards “slow progressors” in the PE-A-treated patients support this possibility. For example, it may be worth investigating whether patients with the different pathogenic genetic variants, and patients at different stages of the disease, respond differently to PE-A
In contrast to the study performed in Spain where the generally safer peripheral venous access 26 was used in all patients, central access was used in almost 42% of patients of the present study. Importantly, this difference did not seem to influence the good tolerability of PE-A procedure with albumin 5%. Overall, the incidences of deaths, severe TEAEs, and AEs leading to treatment discontinuation or study withdrawal were low (only 3 patients accounted for all these events) and were consistent with known TEAEs associated with PE or manifestations of ALS.27,28
It should be noted that, as in the preceding pilot studies, our analyses were limited by a fairly small sample size, and as such may have been underpowered to detect a true difference in ALSFRS-R and FVC scores. In addition, there could be a bias associated with the comparison to controls performed post hoc using a set of ALS patients from an external database to which a theoretical functional decline was assumed. Importantly, however with small pilot studies of this type, measurements of ALSFRS-R and FVC can only be obtained from observed data, so that patients discontinuing related to disease progression or death cannot contribute to change from baseline analyses; given the disposition in this study with 6 discontinuations of 12 participants there may be this type of inherent bias particularly given the contribution of death, AEs, and noting that 2 of the patient-decision withdrawals were prompted by nursing home placement related to disease progression resulting in transportation issues. Larger studies are needed to overcome the high variability in the patients’ response to treatment, likely due to the intrinsic heterogeneity of the disease.
Conclusions
In summary, our results indicate that PE-A with albumin 5% replacement for the treatment of ALS was safe and well tolerated, but it did not demonstrate therapeutic benefit. However, the heterogeneity in the response to treatment confirmed that a number of questions and concerns still persist. A consistent characterization of ALS patients into responder subpopulations would provide a better targeted PE-A approach to ALS treatment. A combined analysis of the results from the Spain and US studies is warranted.
Footnotes
Acknowledgements
Data used in the preparation of this article were obtained from the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) Database. As such, the following organizations and individuals within the PRO-ACT Consortium contributed to the design and implementation of the PRO-ACT Database and/or provided data, but did not participate in the analysis of the data or the writing of this report: ALS Therapy Alliance; Knopp Biosciences; Neuraltus Pharmaceuticals, Inc.; Neurological Clinical Research Institute, MGH; Northeast ALS Consortium; Novartis; Prize4Life Israel; Regeneron Pharmaceuticals, Inc.; Sanofi; Teva Pharmaceutical Industries, Ltd; The ALS Association. Jordi Bozzo PhD, CMPP (Grifols) is acknowledged for medical writing and editorial support in the preparation of this manuscript.
Availability of Data and Materials
Available on reasonable request.
Competing Interests
This study was sponsored by Grifols. EM, MB, MKW, NR, and WL are full-time employes of Grifols, a manufacturer of plasma-derived albumin products. ZS has previously been paid by Grifols, S.A to consult and give lectures on a different study. The remaining authors have no conflicts to declare.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Grifols International, SA.
Consent for Publication
Not applicable.
Ethics Approval and Consent to Participate
The protocol and associated documents were reviewed and approved by the CPHS at Dartmouth-Hitchcock Medical Center (reference number: STUDY00029255).