
Abstract
NPM1+ AML after Essential Thrombocythemia (ET) or Myeloproliferative neoplasms is extremely rare. Only 2 cases have been previously reported after Primary Myelofibrosis. Given the extremely poor prognosis of the Blastic phase of Myeloproliferative neoplasms (MPN-BP), the only curative treatment of these patients is allogeneic stem cell transplantation (allo-SCT) after achieving Complete Response (CR). De novo NPM1+/FLT3- AML is considered a good prognosis entity in which allo-SCT is not contemplated as the first option. A 41 year old diagnosed of ET JAK2 V617F+ 4 years before the diagnosis of AML with normal karyotype and NPM1+/FLT3- was treated with conventional AML induction with Cytarabine and Idarubicine and 3 cycles of high dose Cytarabine. At the diagnosis of AML other 2 mutations were noted: EZH2 and IKZ1. After treatment of AML, NPM1+ clone disappeared, and JAK2 V617F clone reappeared. We opted to treat NPM1+ AML as a de novo AML and we decided to follow up during 2 years without allo-SCT. The patient remains in complete response with NPM1 minimal residual disease negative during the follow up. This case exemplifies the nature of NPM1+ AML secondary to MPD as an extremely sensitive disease to induction therapy plus high dose cytarabine and makes that these type of patients perhaps could be managed without the use of allo-SCT.
Introduction
Myeloproliferative neoplasms (MPN), polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) are chronic hematopoietic disorders characterized by clonal proliferation of mature myeloid elements. The course of these diseases is usually associated to vascular complications, bone marrow failure and transformation in Acute Myeloid Leukemia (AML), which is called Blastic Phase of Myeloproliferative neoplasms (MPN-BP). The frequency of MPN-BP differs between the different entities. It occurs in a 1-2% of the cases of ET, 7% in PV and a 16% of cases of PMF in a period of 15 years. 1 The AML transformation usually is a multistep process associated to secondary mutations including mutations of TP53 2 and is associated to bad prognosis.3,4 We describe a case of ET transformed to an AML with NPM1+/FLT3-ITD-.
Case Report
A 41-year-old man was admitted in our department from another hospital due to the diagnosis of Acute Myeloid Leukemia in May 2018. As previous history, he was admitted in June 2014 in other hospital because of myocardial ischemia, with right coronary disease and he was successfully re-vascularized with 2 stents. The patient in 2014 was a 37-year-old man, smoker without other risk factors. Blood test showed a thrombocytosis (1091 × 1012/L) (Table 1). In peripheral smear, the patient showed giant platelets without erythroblast or dacryocytes. A positive JAK2 V617F mutation was detected in peripheral blood and BCR/ABL was negative. Unfortunately, no bone marrow aspiration or biopsy was performed for unknown reasons in that hospital and, despite this, the patient was diagnosed of ET and treated with low dose aspirin and Hydroxyurea (1 g daily adjusted to leukocyte count). In February 2018, the Hydroxyurea was stopped in order to carry out sperm cryopreservation. In May 2018, the patient developed pancytopenia, with a 35% of blasts in peripheral blood smear (Table 1). No clinical symptoms, hepatomegaly or splenomegaly were present at the diagnosis of AML. Bone marrow aspiration showed 85% of myeloperoxidase-positive blasts corresponding to a M2 morphology and with a cup like nucleus (Figure 1) and bone marrow biopsy was performed, confirming uniform infiltration by AML, with megakaryocytes present, and without evidence of striking fibrosis. The phenotype was myeloid: CD34-, CD117+, CD45d, DR+, CD13+, CD33++, CD38+, CD71+, CD123+. Karyotype was 46XY[20]. Molecular biology tests showed NPM1+ mutation type A (c863_864dupTCTG) with a ratio with ABL control of 123295, FLT3-ITD-, IDH1-, IDH2-, and JAK2 V617F was negative. Using a next generation sequencing (NGS) panel with 40 genes, including JAK2 V617F, TP53, and SF3B1, 3 more mutations were found: PRF8 (C.2052G>T), IKZF1 (C.575T>C), and EZH2 (C.2197T>C).
Hematologic evolution from diagnosis of ET and surveillance of AML minimal residual disease using NPM1 and JAKV617F.

Source: Own elaboration.
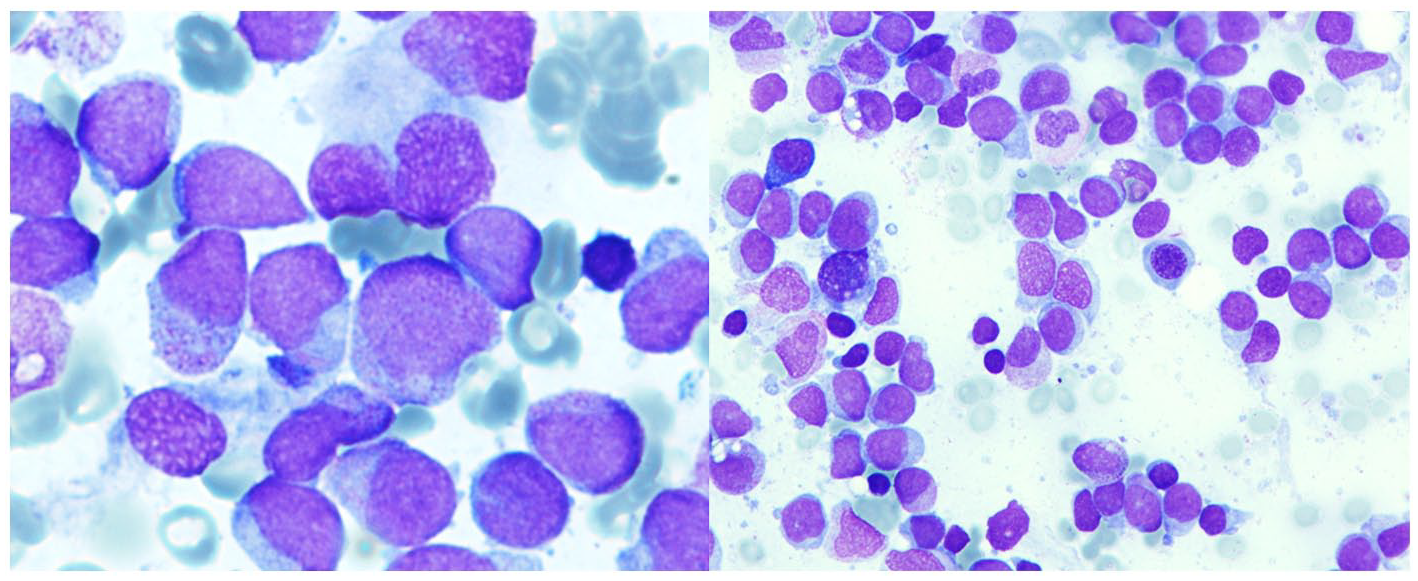
Diagnosis. Pictures of bone marrow aspirate showing massive infiltration by medium-size blasts, with scarce cytoplasm, myeloperoxidase positive blasts corresponding to a M2 morphology and a cup like nucleus. In some of them, we can appreciate Auer rods.
The patient was treated according to PETHEMA 2017 protocol. Induction therapy consisted of Idarubicine 12 mg/m2 × 3 days and Cytarabine 200 mg/m2 × 7 days. He achieved complete remission after first cycle without blasts in bone marrow or peripheral blood, but curiously blood counts after induction showed the typical parameters of a MPN with thrombocytosis (1.5 × 1012/L) and leukocytosis (51 × 109/L) with neutrophilia, and JAK2 V617F mutation was positive again (Table 1). After induction, the patient was treated with 3 consolidation cycles of Cytarabine at dose of 3 g/m2/12 hours days 1, 3, 5. After the first, second, and third consolidation cycles, the peripheral blood counts were absolutely normal (Table 1). Bone marrow aspiration showed myeloid hyperplasia (Figure 2), and NPM1 mutation was negative with positive JAK2 V617F mutation. Ruxolitinib was initiated after third consolidation. Minimal residual disease (MRD) has been followed using real-time quantitative polymerase chain reaction (RQ-PCR) assay for NPM1 mutation and flow cytometry each 3 months and MRD has been persistently negative 3 years after diagnosis. In the other hand, JAK2 V617F mutation remains positive. Control of leukocytosis and thrombocytosis has been achieved with Ruxolitinib, that the patient maintains today at dosage of 20 mg twice daily.
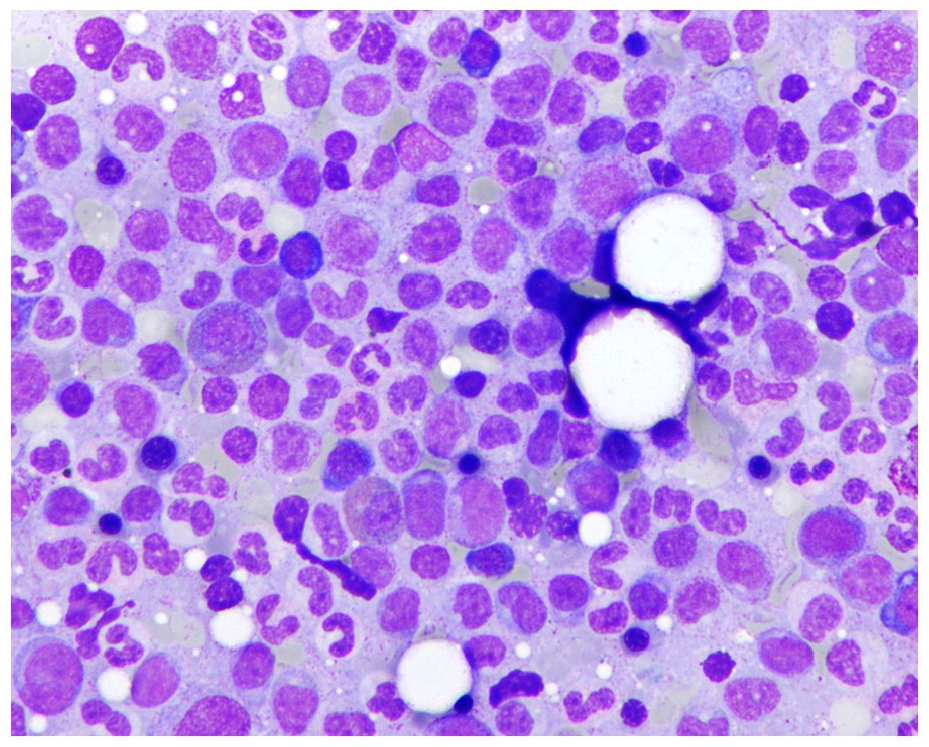
Bone marrow aspirate showing myeloid hyperplasia, after induction therapy.
The patient has a HLA identical sibling. We discussed with the patient and his family the possibilities of treatment after achieving CR. The patient decided not to perform allo-SCT and chose to be followed by MRD, which is monitored every 3 months and remains negative as of today. He signed informed consent for the choice of this follow-up approach.
Discussion
MPN-BP is usually seen as a terminal event with a reported median survival of 3.6 months. 4 The MPN-BP prognosis is related to karyotype, platelet count < 100 × 109/L, transfusion dependence and age.5,6 Clinical factors associated to MPN-BP in ET are low hemoglobin level and the presence of platelets > 1 × 1012/L, and it is independent of JAK2 V617F status. 7 AML transformation has been correlated with the type of treatment (radioactive phosphorus) and not with Hydroxyurea. 8 Given that bone marrow biopsy was not performed at diagnosis of ET, we cannot differentiate between ET or prefibrotic PMF, and a confirmatory initial diagnosis cannot be posed. Prefibrotic PMF is associated with an increased risk of MPN-BP. MPN-BP has been described as a multistep process associated to acquisition of new cytogenetic abnormalities such as −7,−5 or del17p. 9 Even more, TP53 mutation and deletion, TET2, ASXL1, and IDH1 and IDH2 mutations are associated with clonal dominance of JAK2 V617F+ clone and progression to AML. 2 In almost a 50% of the cases of MPN-BF, blasts are JAK2 V617F-.10,11 Treatment consists in conventional 3+7 induction therapy, Azacitidine or supportive care. The rate of complete remission in MPN-BP is around 30%, without differences in overall survival (OS) between conventional therapy, Azacitidine or supporting care. Given the results of conventional chemotherapy in AML secondary to myeloproliferative disease, allo-STC has been assumed as mandatory if the patient achieve CR, with a median survival of 10% at 5 years. 5
NPM1 is one of the more frequently mutated gene in de novo AML, usually associated to normal Karyotype.12,13 In these patients, NPM1+/FLT3- mutation status is associated to good prognosis, and it is recommended that these patients should not be treated with allo-SCT after achieving CR.14,15 NPM1 is considered a founder mutation in the development of AML. This make NPM1 an ideal marker to asses minimal residual disease both in bone marrow 16 or peripheral blood 17 and predict relapse. 18 Otherwise, NPM1 is an uncommon event in Myelodysplastic and Myeloproliferative syndromes. 19 WHO classification excludes NPM1+ AML from the cases of acute myeloid leukemia with myelodysplasia related changes and it is a recognized entity.
We only know 2 cases previously published of NPM1+/FLT3- AML secondary to Primary Myelofibrosis.20,21 In both cases, NPM1+ AML seems to occur independently of the JAK2 V617F+ clone, being similar to NPM1+ AML de novo. 22 There are 3 possibilities that can explain the development of this NPM1+ leukemia in the setting of a previous MPN. Firstly, NPM1+ AML arose independent from the background of a MPN JAK2 V617F+ and can be seen as a de novo AML. Secondly, it could be originated from a pre-JAK2 V617F negative clone common to the ET. The third would be in that a JAK2 V617F-/ NPM1+ clone arose from a JAK2 V617F+ clone by recombination. 21 The concurrent mutation of EZH2 has been described in MPN-BP of ET 23 and secondary AML, 24 but not associated to NPM1+/FLT3-, 2 so in this case could imply the presence of a secondary hit from the ET clone. The mutation in IKZ1 has also been rarely reported in MPN-BP. 25 In our case, JAK2 V617F- AML clone with NPM1 mutation at the diagnosis of ET cannot be ruled out because we have no sample to perform advanced clonal analysis, but the resurgence of ET and the JAK2 V617F clone after AML remission indicates that NPM1+ AML can be an independent event.
The treatment in this patient was a dilemma after the third cycle of consolidation (allo-SCT or surveillance). Generally, MPD-BP is seen as a terminal event that needs treatment with allo-SCT as the only way to cure the disease with a great risk of morbidity and mortality 4 . We opted by monitoring NPM1 MRD to detect preclinical relapse as ELN guidelines recommended.15,26
Conclusion
This case is an example that NPM1+/FLT3- AML independently of the origin of the leukemia clone, must be treated as de novo AML, being of better prognosis than MPN-BD.20,27 Monitoring of MRD permits a tailoring of the treatment and response and permits differing the use of allo-SCT.
Footnotes
Acknowledgements
This case report has been published with the authorization and written consent of the patient here aforementioned.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.