
Abstract
On September 27, 2023, the CureSHANK nonprofit foundation sponsored a conference in Boston, Massachusetts, to identify gaps in knowledge surrounding SHANK3-related epilepsy with the goal of determining future research priorities and recommendations. In addition to patient families and members of the CureSHANK community, participants in the conference included a broad cross-section of preclinical and clinical researchers and clinicians with expertise in SHANK3-related epilepsy as well as representatives from the pharmaceutical industry. Here we summarize the outcomes from comprehensive premeeting deliberations and the final conference recommendations, including (1) gaps in knowledge related to clinical science, (2) gaps in knowledge related to preclinical science, and (3) research priorities moving forward.
Plain language summary
Phelan-McDermid Syndrome, a rare genetic disorder linked to the SHANK3 gene, manifests in a spectrum of clinical phenotypes including intellectual disability, autism spectrum disorder, and epilepsy. Epilepsy has been particularly under-investigated in this syndrome, and most of the animal models studied to date do not display seizures. On September 27, 2023, the CureSHANK nonprofit foundation sponsored a conference in Boston, Massachusetts, to identity gaps in knowledge surrounding SHANK3-related epilepsy. Conference attendees included patient families, basic scientists, clinical researchers, clinicians and representatives from the pharmaceutical industry with interest in SHANK3-related epilepsy. This review summarizes the outcome of this conference, including a summary of current state of knowledge and resources available, gaps in our understanding, priorities for future research in this important manifestation of PMS.
Introduction
SHANK3 is a postsynaptic density scaffolding protein located primarily in excitatory synapses, which are found in abundance on both excitatory and inhibitory neurons. The SHANK3 protein has various roles, including interfacing with signaling molecules, ion channel receptors, and cytoskeletal proteins.1,2 Pathogenic variants in the SHANK3 gene, located on chromosome 22q13.3, are now understood to be associated with a variety of clinical phenotypes including intellectual disability (ID), autism spectrum disorder (ASD), and epilepsy. 3 The resultant syndrome, Phelan-McDermid syndrome (PMS), is the genetic condition caused by SHANK3 haploinsufficiency due to a chromosomal 22q13.3 deletion encompassing SHANK3 or a SHANK3 sequence variant.4–6 Patients with PMS experience diverse neurodevelopmental manifestations including global developmental delay (GDD), ID, ASD, and other behavioral challenges, impaired or limited language, hypotonia, and epilepsy.
Figures on the incidence and prevalence of PMS are limited since there are no birth cohorts or long-term population studies. The incidence of PMS in European countries is estimated to be at least 1 in 30,000. 7 According to Ambry Genetics, SHANK3 pathogenic and likely pathogenic variants are detected at a rate of ~4–5/10,000 (0.05% of patients tested on exome or panels containing this gene) and ~1/1000 for microarray (0.1%) (personal communication, 4/1/24). SHANK3 variants are one of the more common variants among many “synapse development and function” related genes implicated in ASD. 8
The conference members reviewed the literature regarding molecular and cellular roles of SHANK3 protein and the various model systems that have been utilized to investigate SHANK3 deficiency. It is clear from genetic analysis of individuals with PMS that disruption of SHANK3 is sufficient for seizures.6–9 Of note, trisomy of the same region (22q) and interstitial duplications of SHANK3 also cause epilepsy,10,11 further supporting a role for alterations in SHANK3 expression in driving pathophysiology.
CureSHANK is a nonprofit foundation dedicated to accelerating the development of cures and treatments for SHANK3-related disorders through research. On September 27, 2023, the CureSHANK Foundation sponsored a conference at Boston Children’s Hospital in Boston, Massachusetts, to identify gaps in knowledge regarding SHANK3-related epilepsy with the goal of determining future research priorities and recommendations (see Supplemental Table 1 for list of participants in addition to the authors). In preparation for the conference, premeetings were held by both the preclinical and clinical researchers to map out the state of the field and begin to identify knowledge gaps. The conference brought together 24 participants, including patient families and members of the CureSHANK community, a broad cross-section of preclinical and clinical scientists from academia and the pharmaceutical industry, as well as clinicians with expertise in SHANK3-related epilepsy. The conference goals were to identify research priorities and recommendations to define the parameters of CureSHANK’s SHANK3-related epilepsy research commitment. The conference members identified several knowledge gaps in the field of preclinical and clinical research related to SHANK3-related epilepsy. Here we summarize the outcomes from the comprehensive premeeting deliberations and final conference recommendations, including (1) gaps in knowledge related to clinical science, (2) gaps in knowledge related to preclinical science, and (3) research priorities moving forward.
Body
Current knowledge: Clinical science
The state of current knowledge was evaluated by reviewing the literature of studies discussing the prevalence or characteristics of epilepsy in individuals with PMS. Studies to be reviewed were identified by performing a search of the PubMed database using the key terms “Phelan-McDermid syndrome” and “epilepsy.” Studies reporting only case reports were excluded. Thirteen retrospective studies were identified that had any discussion of epilepsy prevalence or characteristics and one prospective study. 12 In addition, one study reported the prevalence of epilepsy specifically in individuals with point mutations in the SHANK3 gene. 6 The prevalence of seizures from these studies was between 14% and 70% with the prospective study reporting a prevalence of 41%. 13 Detailed descriptions of seizure types, frequency of seizures, and medication usage were not reported in most of these studies. One study did comprehensively report seizure types present in PMS at one site. 14 They reported a wide range of seizure types from focal, atonic, generalized tonic-clonic, and myoclonic. In addition, they found that 20% of individuals with PMS had at least one-lifetime episode of status epilepticus, and 18% had a diagnosis of Lennox Gastaut syndrome (LGS). At least two studies further reported frequent abnormalities in electroencephalogram (EEG) including in PMS individuals without a history of clinical seizures.12,14
Gaps in knowledge: Clinical science
The conference members identified five major knowledge gaps in the field of clinical science related to SHANK3-related epilepsy.
Why do only a minority of individuals with SHANK3 pathogenic variants develop epilepsy?
In individuals with PMS, the prevalence of epilepsy ranges from 25% to 30% with about half of patients developing at least one single lifetime seizure.6,12,14,15 Reported seizure types include atypical absence, atonic, tonic, tonic-clonic, and myoclonic seizures. Age of seizure onset, seizure frequency, and seizure burden are all highly variable. To date, there is no clear genotype-phenotype correlation; an increase in the size of the deletion is not related to the presence or the severity of epilepsy.3,9 If we could better understand why certain patients develop seizures, we may have the opportunity to target potentially modifiable risk factors.
What is the prevalence of refractory epilepsy in this population? What is the natural history of refractory epilepsy in this population?
Although we have at least 12 retrospective studies describing the prevalence of seizures in individuals with PMS, we do not have a detailed description of seizure severity or of natural history in this population. A clinical review of 33 patients with PMS and epilepsy at Texas Children’s Hospital described a broad range of seizure burdens—from one seizure to hundreds per day. 14 Twenty percent of these patients suffered from at least one episode of status epilepticus and eighteen percent of these patients were diagnosed with LGS, a severe epileptic encephalopathy. If we could better understand why certain patients develop severe refractory epilepsy—perhaps due to environmental or genetic “second hits,” and whether and how epilepsy changes over time, we may have the opportunity to better screen for and even prevent this progression.
Are there any mechanisms to predict which patients are at highest risk to develop epilepsy? Is EEG predictive?
At this time, we are unable to predict which patients will develop epilepsy based on genotype, clinical presentation, biomarkers, or scalp EEG. Indeed, there is a range of EEG findings in PMS: a majority of patients do have generalized slowing and/or epileptiform discharges, including spikes during sleep, but these findings are often non-specific and do not predict the presence or absence of epilepsy. 12 In the absence of a predictive model for epilepsy, several labs have turned to using quantitative EEG or other functional readouts to study any electrophysiological differences between the brains of individuals with PMS and typically developing controls. 16
Can we identify any structural differences in the brains of individuals with SHANK3 mutations and epilepsy versus those without?
Comparative neuro-imaging data is lacking and could present an opportunity for early identification of patients who are at risk for the development of epilepsy. More complex imaging modalities such as volumetric MRI may be useful in this space. Detailed analysis of post-mortem brain tissues could also enhance our understanding of the pathobiology of SHANK3-related epilepsy.
Are there certain anti-seizure medications that are particularly effective for this population? Conversely, are there certain anti-seizure medications to avoid in this population?
Within the Texas Children’s population, patients were prescribed sixteen different anticonvulsant medications, but none with clear superiority. 14 Levetiracetam in particular has been identified as an anti-seizure medication with considerable efficacy for some PMS patients, yet it can trigger significant mood and behavioral side effects for others. With more detailed evaluation of medication response in this population, it may be possible to offer patients the most effective anti-seizure medication(s) with the least debilitating side effects.
Gaps in knowledge: Preclinical science
What role do different isoforms of the SHANK3 gene play in the brain?
There is no definitive gene model for SHANK3 expression, and most of the information in databases is pieced together from old sequencing data. SHANK3 is a complex gene of exceptional length, with multiple intragenic promoters. Factors that regulate its expression are not entirely understood. The gene is in a GC-rich region, making analysis difficult. However, combination of long-read sequences and high coverage in single-cell RNA sequencing can help develop a definitive gene model, with specificity for different cell types and developmental stages.
Where in the brain is SHANK3 expressed?
SHANK3 clearly plays a role at sites postsynaptic of excitatory synapses in many neuronal subtypes, including inhibitory neurons. However, we do not yet understand which neurons and which brain regions are most involved, or whether the function of certain cell types is more sensitive to loss of SHANK3. Differences in expression patterns across species may play a role in the differential manifestation of epilepsy, highlighting the need for detailed profiling of expression patterns from animal models to humans. Studies in mice have demonstrated a role of Shank3 deficiency in inhibitory neurons in hyperexcitability,17,18 but further work in this area using deletion of the whole gene was deemed a priority.
Which is the best mouse or rodent model of Shank3 loss for understanding epilepsy?
SHANK3 isoforms in brain lysates are largely conserved between mice and humans, 19 and several distinct genetically modified mouse models have been generated impacting different isoforms or to introduce specific mutations (Table 1). Where studied, genetically modified mouse and rat models display deficits in synaptic plasticity, including in the heterozygous state.20–22 However, none of them display spontaneous seizures. Crossing the knockout allele into different genetic backgrounds does not appear to change the phenotype. 23 In fact, knockouts appear to be resistant to PTZ-induced seizures, 24 consistent with reduced glutamatergic tone, while in others kainic acid-induced seizures appear to be increased. 25 The consensus at the workshop was that mouse and rat models can still be useful and that the preferred mouse models are those that remove all isoforms of the Shank3Δ4-22, gene (Shank3flox4-22, Jax Strain #032169; Strain #032158).20,23 There was also discussion on moving away from the emphasis on clinical seizures in rodents and start to investigate other electrophysiological findings in both preclinical and clinical studies, such as interictal discharges or atypical cross-frequency coupling, which has been detected in clinical studies. 16
Mouse, rat, and human iPS cellular models of PMS.
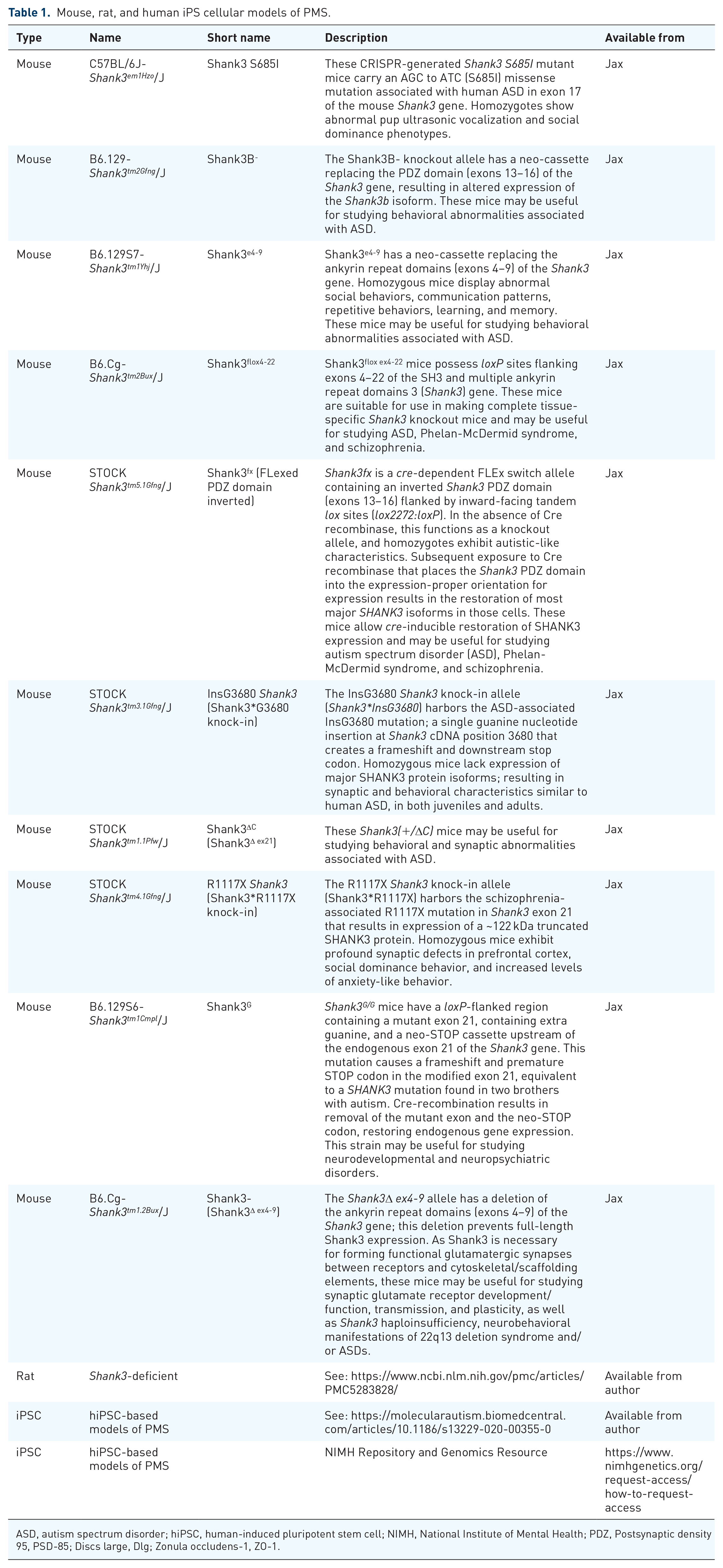
ASD, autism spectrum disorder; hiPSC, human-induced pluripotent stem cell; NIMH, National Institute of Mental Health; PDZ, Postsynaptic density 95, PSD-85; Discs large, Dlg; Zonula occludens-1, ZO-1.
What can we learn from non-human primate models of SHANK3?
Since rodent models do not display spontaneous seizures, there has been increased interest in developing non-human primate models, such as macaques and marmosets.26–28 There have not yet been significant advances in understanding epilepsy using these models, although they are recent and rare models. More in-depth studies of these models using EEG monitoring will be necessary to understand how well they mimic human disease.
What can we learn from human neurons in culture?
Several studies have turned to induced pluripotent stem (iPS) cell models (Table 1) to investigate the role of SHANK3 protein in human neurons and have identified transcriptional and synaptic deficits.29–31 Interestingly, initial studies using mixed excitatory and inhibitory neuron cultures indicate early maturation and hyperexcitability 32 and require follow-up with additional iPSC lines. Further work using different 2D and 3D differentiation methods is needed to further understand the utility of iPS cell-derived neuronal cultures as a model system to study SHANK3-associated epilepsy. Multiple studies detected elevated intrinsic excitability in SHANK3-deficient human neurons and organoids,29,30,33,34 which may be associated with elevated excitability in neural networks and seizures. Previous studies also reported that heterozygous and homozygous SHANK3 mutations impaired hyperpolarization-activated cation (Ih) channels.33,35 Finally, recent studies reporting transplantation of iPS-cell-derived neurons into rodents open another opportunity to investigate SHANK3 loss. 36
How do neuro-inflammation and/or immune dysregulation interact with SHANK3?
SHANK3 is expressed in the spleen at high levels, suggesting some interaction with the immune system. However, there have not been any studies pursuing this line of work, despite a tight link between epileptogenesis and inflammation. In parallel, investigation of inflammatory markers in the blood and in the brain may be helpful to further gauge the importance of neuro-immune interactions in PMS.
Research priorities
The conference members identified two high-priority research areas in the field of SHANK3-related epilepsy moving forward, with the goal of expediting progress in our understanding and treatment of SHANK3-related epilepsy (Table 2). In addition, workshop participants highlighted the importance of independent replication of important findings to ensure that the path toward treatment is based on rigorous reproducible results.
Summary of high-priority research areas in the field of SHANK3-related epilepsy.

Priority area 1 (Clinical Science): Understand the natural history of epilepsy in patients with SHANK3-related epilepsy
With a more detailed characterization of epilepsy presentation and progression in patients with a pathogenic variant in the SHANK3 gene, we may be able to identify “second hits” such as genetic markers, environmental triggers, neuro-immune interactions, and/or other illnesses that contribute to epileptogenesis. A large NIH-funded multi-site natural history study is ongoing and collecting data across many domains (Developmental Synaptopathies Consortium, NCT02461420, See Table 3). We may be able to utilize such existing cohorts to interrogate this question, and we encourage the use of epilepsy as a secondary outcome in future trials. However, one immediate priority would be to develop a registry of patients with refractory epilepsy to better study both the genetic and non-genetic contributors to exact differences between this particular cohort and other individuals with PMS without severe epilepsy.
List of assessments performed in the Developmental Synaptopathies Consortium for PMS participants.
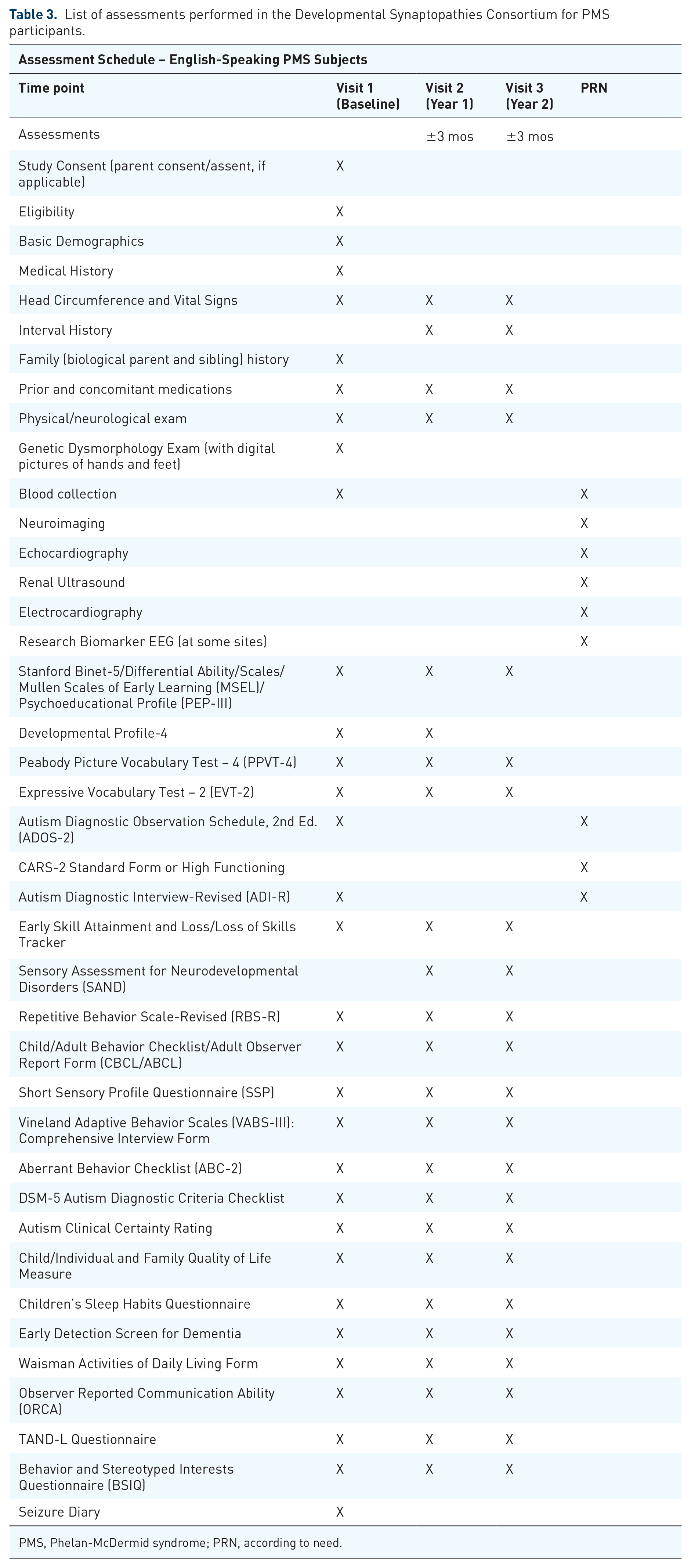
PMS, Phelan-McDermid syndrome; PRN, according to need.
Priority area 2 (Preclinical science): Create a model of epileptogenesis for SHANK3-related epilepsy
None of our rodent models have spontaneous seizures; exploring subclinical electrophysiological changes in both clinical and preclinical studies will help understand their relationship with seizures. There needs to be more work on non-human primate models as well as with iPS-cell-derived neurons to ask whether one can learn more about epileptogenesis in those platforms. For instance, generating human neurons from PMS patients with and without epilepsy may allow comparison of excitability in culture. Understanding the neurophysiology underlying electrographic changes such as cross-frequency coupling may provide clues to mechanisms that are vulnerable to SHANK3 loss, such as feedback inhibition. These more subtle manifestations of hyperexcitability may give clues about the origins of seizures and can be probed with modern circuit neuroscience tools.
Conclusion
This manuscript provides a narrative review of the state of the resources available in PMS to address epilepsy associated with this syndrome. The limitations of our approach include the fact that we did not perform an exhaustive scoping review of the literature, especially in terms of preclinical models of PMS. We also did not perform a formal Delphi method to reach a consensus on the priorities. Despite its shortcomings, this workshop was the first step to summarize the resources available, gaps in knowledge, and major priorities for research in this previously under-investigated topic. Better characterization of the natural history of epilepsy is absolutely necessary both to understand the process of epileptogenesis in PMS as well as to develop clinical trials that address epilepsy as either a primary or secondary outcome major. Another major priority—to develop models of epilepsy in either rodents, non-human primates, or human cell-based cultures—will be crucial to both identifying mechanisms and treatment targets but also potential translatable biomarkers for clinical trials.
Supplemental Material
sj-docx-1-trd-10.1177_26330040241273464 – Supplemental material for A roadmap for SHANK3-related Epilepsy Research: recommendations from the 2023 strategic planning workshop
Supplemental material, sj-docx-1-trd-10.1177_26330040241273464 for A roadmap for SHANK3-related Epilepsy Research: recommendations from the 2023 strategic planning workshop by Margaret C. Savage, Geraldine Bliss, Joseph D. Buxbaum, Jordan S. Farrell, April R. Levin, Siddharth Srivastava, Elizabeth Berry-Kravis, J. Lloyd Holder and Mustafa Sahin in Therapeutic Advances in Rare Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.