
Abstract
Persistent Mullerian Duct Syndrome (PMDS) is an extremely rare disease with less than 300 cases recorded in medical literature. Our patient was a 37 year old male who presented at the medical office with hematospermia as his sole complaint. He had previously undergone left orchidopexy and presented with hypotrophic left testicle and right testicle agenesis. PMDS differential was considered with the clear observation of a uterus-like structure during pelvic ultrasonography. The organs were later studied in magnetic resonance imaging and confirmed by post-surgery anatomopathological examination. Patient was discharged 24 h after surgery and developed azoospermia post-surgery.
Plain language summary
PMDS is a disease which has less than 300 cases in medical literature. It is a congenital condition characterized by the development of female genital organs such as the uterus and ovaries, in an otherwise normal male individual. The fetal development of these structures begins when the male fetus develops his genitalia, during the period when he must produce a hormone (anti-mullerian hormone), which suppresses female genitalia growth. Since this fetal stage is the turning point for genital development, lack of this hormone commonly results in the presence of functional female genital organs in an adult male, which characterizes the syndrome. Multiple reports also associate the syndrome with ectopic testis (cryptorchidism) or gonadal absence and dysfunctional sexual cell production.
The aim is to present a rare presentation of an already extremely rare disease in order to enrich the literature with another case of PMDS and the outcome of surgical correction.
After discovering female organs in the male pelvis during an ultrasound scan, an elective surgery was performed to evaluate the removal of the uterus, fallopian tubes, ovary and vaginal canal through video laparoscopy.
The overall medical knowledge about PMDS is rather limited due to the reduced number of cases and the relatively wide variety of presentations. This article is useful to present a rather rare presentation, in which cryptorchidism and testicular agenesis were concomitant with hematospermia. Other than that, the diagnosis was done late in the patient’s life, having lived over three decades with female genitals in his pelvis without any malignant (cancerous) mutations. The case report can also provide a record for the outcome of azoospermia, which is the absence of motile (and hence viable) sperm in the semen, following a non-complicated post-surgical recovery, which suggests unknown mechanisms may be involved in gonadal development after birth, and a different endocrine balance in patients with the syndrome.
Introduction
The Persistent Mullerian Duct Syndrome (PMDS) is a very rare phenomenon with less than 300 cases described in literature. 1 It affects phenotypical males with 46 chromosomes (XY karyotypes) who develop anatomically variable female internal organs, which are Mullerian derivates. 2
The organogenesis of the male external genitalia is not affected since testosterone levels are usually normal. The female organ development in a male individual occurs because of a defect in genes coding for the Anti-Mullerian Hormone (AMH) located at 19p13, which is produced by Sertoli cells in the normal male, or due to a defect of its type II receptor (AMHR-II) located at 12q13, accounting for the regression of Mullerian structures, which usually happens as soon as the 8th week of pregnancy. 3
In patients with PMDS, instead of having Mullerian structures reduced to the normal male’s prostatic utriculus, a normal uterus, fallopian tubes, both ovaries and the proximal third of the vagina are present and relatively well-developed, sometimes with functional sexual maturation. In even rarer cases, PMDS is associated with transverse testicular ectopia and diagnosis is established when Mullerian duct structures are discovered in ultrasonography or surgical exploration of common associated conditions and symptoms, which is what happened with the case described in this report.4,5
Case review
Our patient (MSF) was a 37-year-old male who presented at the medical office with the sole complaint of hematospermia of an undetermined cause. He was married and carried a normal sexual life, but had no children. His only relevant medical record accounted for bilateral inguinal herniorrhaphy and left orchidopexy during infancy.
During the physical examination, the right testicle was not palpable, and the left testicle was hypotrophied. Pelvic ultrasonography and laboratory tests were performed to evaluate hematospermia, such as spermogram, coagulation proofs and urinalysis. He was then submitted to an ultrasonographic examination by the same assisting physician (Dr. Iwens) which revealed a hypoechoic tubular structure, measuring 56 mm × 29 mm × 18 mm with a volume of 15.7 cm3 and resembling the uterus.
The hypothesis of Mullerian malformation was then considered. Magnetic resonance imaging of the pelvis was conducted (Figure 1) and a structure resembling a rudimentary uterus with vaginal canal and ovary was described by the radiologist. Surgical indication was promptly established to perform the total hysterectomy, which was done via video laparoscopy with a total surgical time of 70 min and blood loss of less than 50 ml.
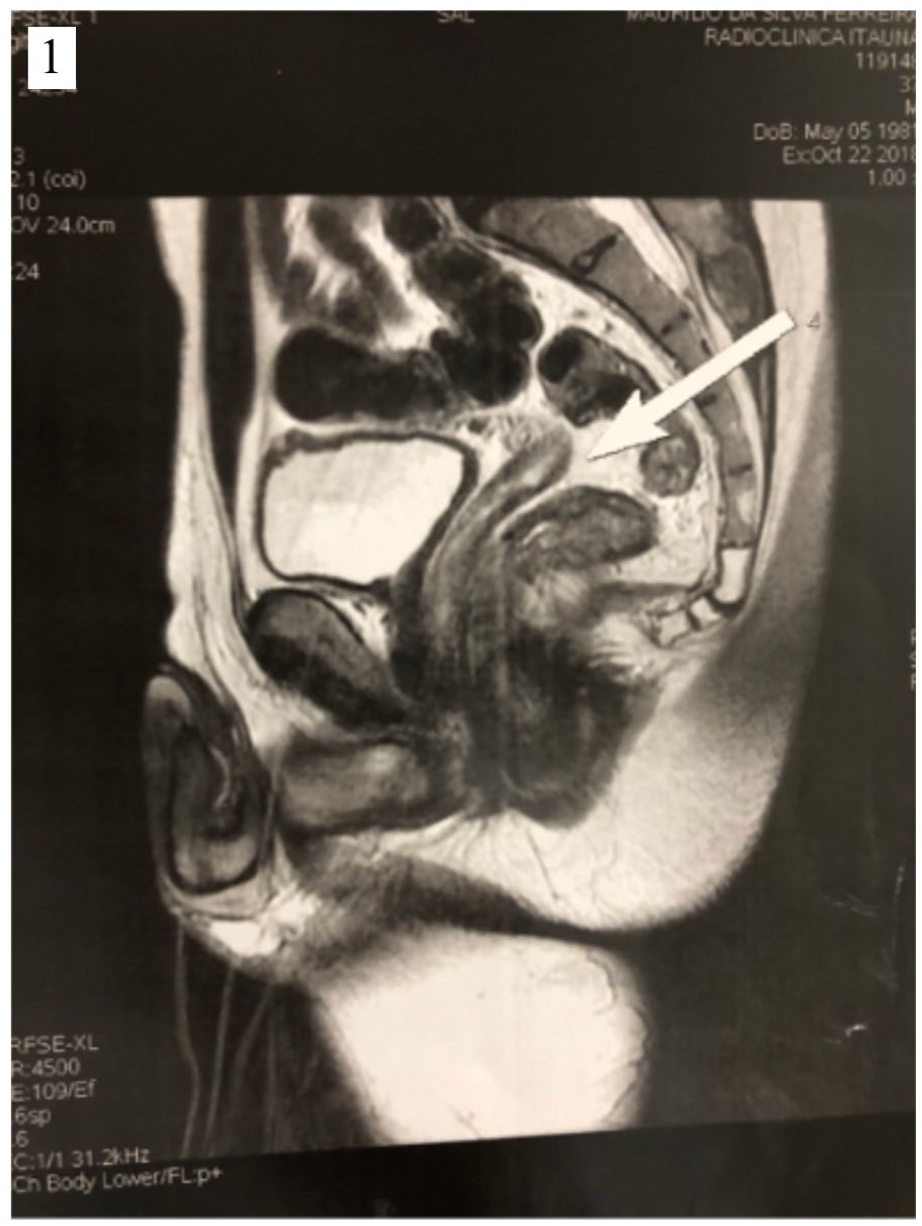
Pelvic MRI in T2 showing a solid organ between the bladder and the rectum, with the usual appearance of the anteverted uterus. The arrow points to its posterior end.
The preferred surgical approach is a laparoscopic radical hysterectomy (see Figures 2–5), which contemplates the removal of the uterus, fallopian tubes, the only formed right ovary, cervix, vaginal canal and appendages, which were later confirmed by anatomopathological examination and showed no signs of cellular atypia or neoplastic transformations. The uterus had a normal pyriform appearance and size (as one would expect to find in a nulliparous female patient) and was found in a low retroperitoneal site, attached by fibrous and connective tissue to the posterior muscular abdominal wall. The fallopian tubes were also anatomically normal. The right ovary found was apparently slightly smaller than a same age woman’s would be, although precise ultrasound measurements were unfortunately lost. Cystoscopy was performed during surgery, showing no communication between the vaginal canal and the urethra. The patient recovered satisfactorily post-operations and was discharged 24 h later. He returned to work after 15 days and had no complaints in performing usual activities. Unfortunately, he then developed azoospermia (absence of sperm).

Laparoscopic approach to the uterus site. The uterus corpus (indicated by the black asterisk) is seen as eutrophic, attached to the abdominal cavity through a peritoneum-like membrane. The left fallopian tube is also visible (indicated by the solid arrow), including attached yellowish adipose tissue.

Following the cervix, an incision was performed in the membrane to expose the vaginal cul-de-sac (indicated by the black star), which is grappled by the surgeon.

Soon after exposing the vaginal cul-de-sac. Because of the dissection, it is possible to determine its limits and proceed with extirpation. The vaginal canal is then extorted from the attached connective tissue and removed.

Exploring the dissected vaginal canal to determine its end (cul-de-sac). The whitish fibrous tissue seen (indicated by the dotted arrow) represents a fusion of connective tissue, resembling fibrosis, and closes the vaginal canal where the vulva should be in women.
Discussion
The genitourinary tract development involves three stages: pronephric, mesonephric and metanephric. Internal genitalia originates from the mesonephric ducts. At this stage, mesonenephric ducts are called Wolffian ducts. 5 Mullerian and Wolffian ducts are usually both present in the fetus at 7 weeks of gestation.
Male sex differentiation is driven by two distinct hormones and each one is produced by a separate cell compartment of the fetal testis. The first hormone is testosterone, which is produced by fetal Leydig cells and promotes the differentiation of the Wolffian ducts into the epididymis, vas deferens and seminal vesicles. The second hormone is the AMH, which is a member of the TGF-β family and is synthesized by fetal Sertoli cells. AMH is responsible for the regression of fetal Mullerian ducts, which would differentiate into the uterus, fallopian tubes and upper vagina if this hormone is absent. 2 Also another hormone plays a role in genital development: dihydrotestosterone. Dihydrotestosterone induces differentiation of external genitalia, which explains why the external structures are usually preserved in PMDS patients. 4
Due to a failure of AMH synthesis, release or action, PMDS patients have both Wolffian and Mullerian ductal structures, but normally developed male external genitalia and Wolffian structures. 4 The development of Mullerian derivatives is extremely variable. The uterus may be comparable in size to that of normal females, but it is often much smaller. 2
AMH gene mutation (PMDS type 1) is responsible for approximately 45–52% of genetically proven PMDS cases. AMH receptor gene (AMHR2) mutations (PMD type 2) are responsible for the remaining 40% of cases.3,4 Patients with AMH or AMHR2 gene mutation show no differences in the anatomic phenotype.
In males, serum levels of AMH usually remain high until 2 years of age and are measurable until puberty, before decreasing to undetectable levels in the adult male. Low or undetectable AMH levels in PMDS cases indicate AMH mutation, whereas high AMH levels may indicate mutation in the AMHR2 gene. 5
Concerning treatment, each presentation would require individual evaluation, but it tipically involves removing the female organs entirely.6 –8 Laparoscopy is considered the gold standard to perform the extirpation.9,10 The main concern of surgeons seems to be the greater size and unusual vascularization patterns of Mullerian structures in PMDS patients. 8
Conclusion
The first published case report on PMDS dates back to 1939, which was published by Alfred Jost. That report has great scientific meaning, showing for the first time that sexual differentiation is directly determined by two testicular hormones that vary in origin and function. These hormones are testosterone secreted by Leydig cells, and AMH which is secreted by Sertoli cells.
The syndrome is considered to be rather rare, accounting for only 150 cases reported in literature by 1993, although the number of cases are gradually increasing.
The prevalence of the condition can be underrepresented because few healthcare providers know about it; it is often diagnosed by accident during an imaging procedure or during surgery. Once the diagnosis is established, the orchidopexy can be performed to preserve the patient’s fertility and to monitor inguinal testis.
PMDS shows an overall good prognosis despite the high infertility rate observed in patients, as well as Mullerian and testicular degeneration. This is why knowing that the syndrome exists and sometimes presents with subtle findings can be the key to reach the best possible results from the condition management.