
Abstract
Mucopolysaccharidoses (MPS) are a group of inherited lysosomal storage disorders caused by deficient levels and/or activity of glycosaminoglycan (GAG)-degradative enzymes. MPS are characterized by accumulation of the mucopolysaccharides heparan sulfate, dermatan sulfate, keratan sulfate, or chondroitin sulfate in tissues. We report the case of a 38-year-old woman with a history of joint restriction and retinitis pigmentosa who developed bivalvular heart failure requiring surgery. It was not until pathological examination of surgically excised valvular tissue that a diagnosis of MPS I was made. Her musculoskeletal and ophthalmologic symptoms, when placed in the context of MPS I, painted the diagnostic picture of a genetic syndrome that was overlooked until a diagnosis was made in late middle age.
Plain Language Summary
• A 38-year-old woman with heart failure had heart valve surgery. Examining her cardiac valve tissue under the microscope suggested a metabolic disorder called mucopolysaccharidosis type I (MPS I).
• MPS I is due to defective breakdown of sugar molecules (called glycosaminoglycans or GAGs for short) in the body which then can accumulate, causing dysfunction.
• Our patient had short stature, a curved spine, stiff joints, and a degenerative eye disease called retinitis pigmentosa, all of which were due to her undiagnosed MPS I.
• Most patients with MPS I are discovered on newborn screening when they are babies, or at very young ages due to severe symptoms related to the disease.
• Our patient had a form of MPS I that was less severe, and the first symptom she received medical care for was her eye symptoms.
• A diagnosis of MPS I made in middle adulthood is unusual for MPS I, and so is an important learning case for providers as there were clues hidden in her medical history that suggested a genetic or inherited syndrome.
• Our genetics specialists were able to make a definitive diagnosis of MPS I and begin treatment with enzyme replacement therapy, as well as provide information for the patient about her risk of passing this disease on to children.
Introduction
The mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders caused by deficient levels and/or activity of glycosaminoglycan (GAG)-degradative enzymes.1,2 Defective enzymatic breakdown of GAGs results in accumulation of the mucopolysaccharides heparan sulfate, dermatan sulfate, keratan sulfate, or chondroitin sulfate in tissues.1,2 The clinical phenotype of each MPS is determined by the specific enzyme deficiency, the severity of the enzyme deficiency, the target GAG of the deficient enzyme, and sites of accumulation of GAGs in tissues.1,2 We followed the CARE Guidelines when preparing this case report.
Case report
A 38-year-old occupational therapist presented with an acute onset of shortness of breath. Echocardiographic assessment showed severe calcific aortic and mitral valve stenosis, atrial enlargement, pulmonary hypertension, and tricuspid, aortic, and mitral valvular regurgitation with a preserved ejection fraction of 64%. She did not have a history of prior chest radiation or rheumatic fever.
The patient’s prior medical history was notable for hyperopia, congenital hip dysplasia, an umbilical hernia, obstructive sleep apnea, tracheal stenosis causing difficult intubations, carpal tunnel syndrome, underdevelopment of the hand musculature, as well as joint contractures and stiffness beginning in childhood. She was diagnosed with retinitis pigmentosa at age 34 years with evidence of pigmentary retinopathy on examination after noting over a decade of progressive nyctalopia. Chromosomal microarray (CMA) performed at the time of diagnosis of retinitis pigmentosa was negative for duplications or deletions. Family history is unremarkable for cardiac, musculoskeletal, or retinal disease. She had two uncomplicated pregnancies at ages 31 and 34, with two phenotypically normal children. She has a biologic brother who has no health issues. The patient is of normal intelligence and sound mind, with a very successful and fulfilling career as an occupational therapist.
On physical examination, the patient was noted to have macrocephaly (head circumference 60 cm; 99th percentile) and a prominent forehead without coarse facial features, a low nasal bridge, micrognathia, dental crowding, and misaligned teeth with a prominent square jawline (Figure 1(a)). She was of short stature (height 61 inches; 9th percentile), had valgus knee deformity with leg length discrepancy, significant muscle wasting of the bilateral upper and lower extremities, and bilateral thenar atrophy (Figure 1(b)). She also had notable joint contractures of the elbows, wrists, hips, and knees. Spinal examination was notable for scoliosis with spine imaging showing thoracolumbar scoliosis, narrowing of the lumbar spaces, anterior subluxation of L5 onto S1, and dysplastic pelvic bones (Figure 1(c)).
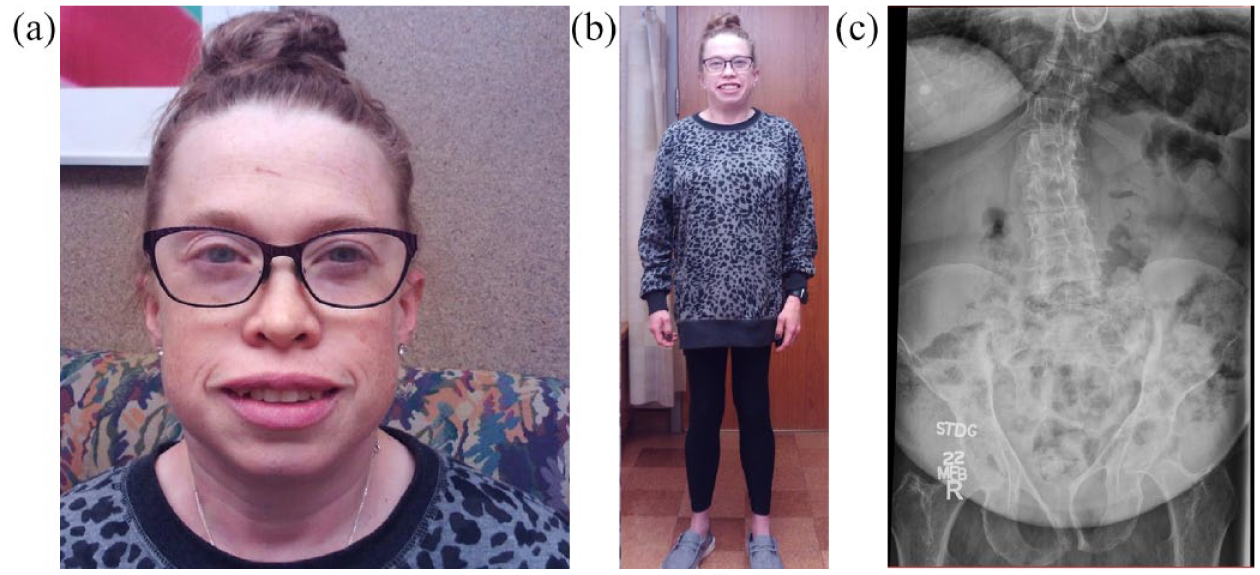
(a) Photograph of the patient’s facial structure indicating macrocephaly, prominent forehead, low nasal bridge, micrognathia, dental crowding, and square jawline. (b) Photograph showing the patient standing. (c) Anterior–posterior radiograph of the thoracolumbar spine shows thoracolumbar scoliosis, narrowing of lumbar interspaces, spondylotic subluxation of L5 on S1, and dysplastic pelvic bones.
Ophthalmologic examination showed bilateral arteriolar attenuation and bone-spicule-like pigmentary changes anterior to the inferotemporal arcades (Figure 2(a)). Optical coherence tomography of the macula revealed extensive loss of the outer nuclear layer, external limiting membrane, and the ellipsoid zone that spared the central 1 mm (Figure 2(b)). Fundus autofluorescence imaging revealed an elliptical ring of hyperautofluorescence 1.5 mm × 2 mm in size centered on the fovea. The foveal avascular zone was also slightly hyperautofluorescent (Figure 2(c)). Visual field testing revealed bilateral ring scotomas though central visual acuity was 20/20 in both eyes (Figure 2(d)). These retinal findings were consistent with retinitis pigmentosa. The remainder of the patient’s eye examination was normal including intraocular pressures, extraocular movements, corneal clarity, and optic disc appearance.

(a) Montage color fundus photograph of the right eye showing attenuated retinal arterioles, a blunted foveal reflex, and a small amount of bone-spicule-like pigmentation anterior to the inferotemporal vascular arcade. (b) Spectral domain optical coherence tomogram (OCT) of the right macula showing extensive loss of the outer nuclear layer, the external limiting membrane, and the ellipsoid zone that spares the central 1 mm. A trace amount of cystoid macular edema can be seen in the inner nuclear layer. Imaging obtained of the left eye, although not included, showed similar changes to the right eye. (c) Fundus autofluorescence image of the right eye showing an elliptical ring of hyperautofluorescence 1.5 mm × 2 mm in size centered on the fovea. The foveal avascular zone is also slightly hyperautofluorescent. (d) Goldmann visual field of the right eye shows a ring scotoma to the I4e isopter. The I2e isopter is constricted to less than 20°.
Electron microscopy of the mitral valve biopsies showed numerous clear vacuolated cells with clear, flocculent material and inclusions in the leaflets and chordae tendineae (Figure 3(a) and (b)). Light microscopy of her native mitral valve showed collections of abnormal vacuolated histiocytes in both the fibrous and spongy layers (Figure 3(c) and (d)). These ultrastructural findings were consistent with mucopolysaccharidosis.
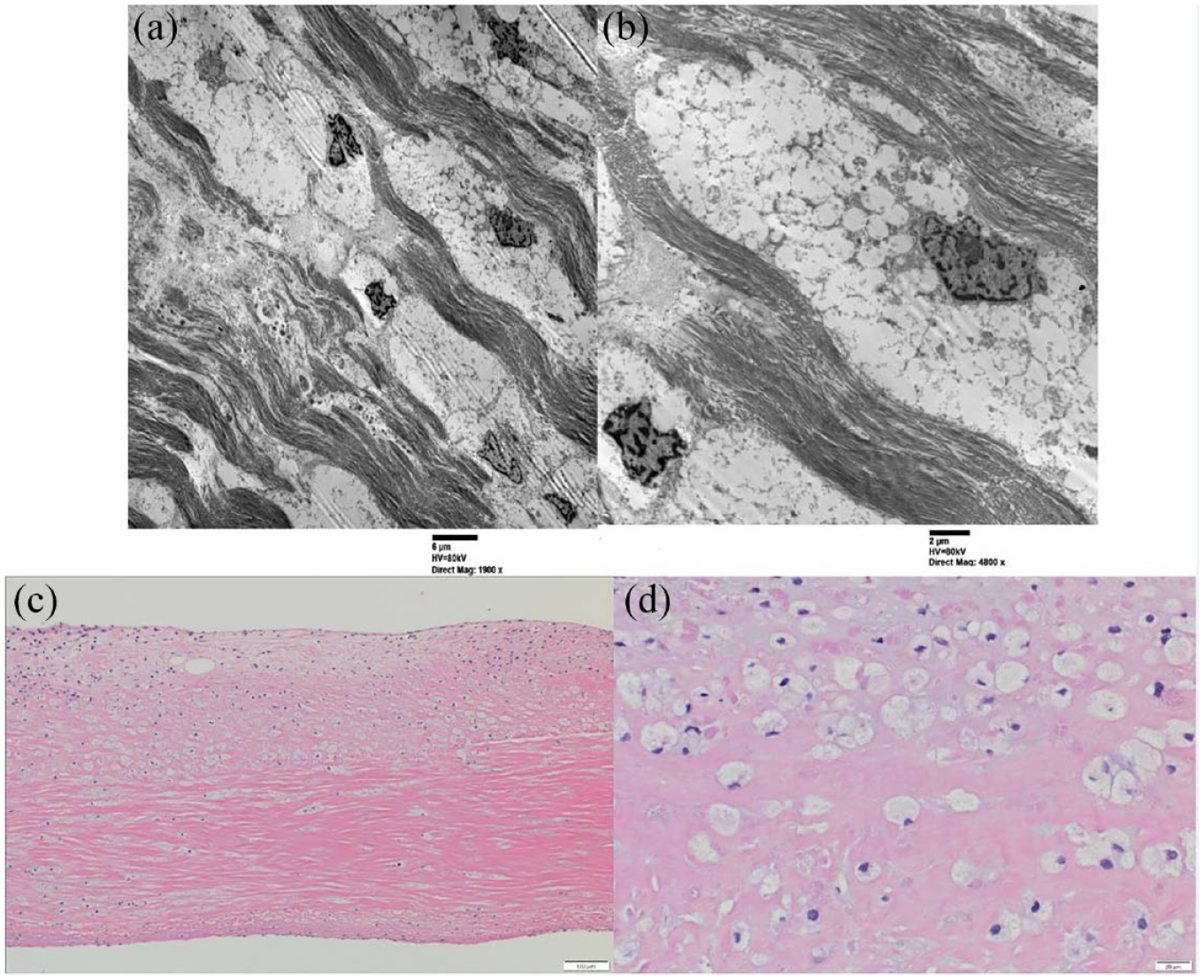
Surgical microscopy specimen of the mitral valve as seen using electron microscopy. Abnormal vacuolated cells with flocculent cytoplasmic material viewed at both (a) low and (b) high magnification. Surgical microscopy specimens of the mitral valve as seen using light microscopy. Collections of abnormal vacuolated histiocytes are present within the fibrous and spongy layers of the mitral valve seen at both (c) low and (d) high magnification using hematoxylin–eosin stain.
CMA and whole genome sequencing analyses were facilitated by clinical genetics. CMA was performed by Mayo Clinic Laboratories in Rochester, Minnesota and used copy number and single-nucleotide polymorphism (SNP) probes on a whole genome array Applied Biosystems (Affymetrix, Waltham, MA, USA) CytoScan HD platform. CMA showed no clinically relevant copy number changes or regions with absence of heterozygosity. Whole genome sequencing performed at GeneDx Laboratories in Gaithersburg, Maryland was positive for two heterozygous variants in the IDUA gene encoding iduronidase. The first variant, c.228 T>A (p.Tyr76Ter) is paternally inherited and classified as pathogenic. The p.Tyr76Ter variant is a nonsense variant predicted to result in protein truncation or nonsense-mediated decay; it has not been previously published as pathogenic or benign to our knowledge and has not been observed at significant frequency in large population cohorts. 3 The second variant, c.1577T>C (p.Leu526Pro), is maternally inherited and classified as likely pathogenic. The p.Leu526Pro variant was observed to be in trans with the paternally inherited p.Tyr76Ter variant. In silico analysis supports a deleterious effect for the maternally inherited p.Leu526Pro variant.
Biochemical testing identified low-serum alpha-
Upon genetics consultation, and due to the atypical presentation of symptoms in this case, informed consent was obtained for clinical photography and case report publication.
Discussion
In this case report, we present a 38-year-old woman with severe cardiac valvular dysfunction out of proportion to her age, retinopathy, and musculoskeletal comorbidities that collectively raised concern for an underlying genetic condition. The surgical pathologic findings were suggestive of a metabolic storage disorder in her diseased valves.1,2,4 Identification of bi-allelic pathogenic variants in the IDUA gene, followed by biochemical testing showing reduced iduronidase activity, led to the correct diagnosis of MPS I for this patient.1,2 The novelty of this case presentation is based on the attenuated phenotype and atypical presentation.
There are a number of instructive points in the discussion of this case. The severe end of the spectrum (Hurler syndrome) is readily recognizable. However, more attenuated disease (Hurler–Scheie and Scheie syndrome), as exemplified in this case report, can be a diagnostic challenge. Symptoms in attenuated MPS I are slowly progressive, with diagnosis typically in mid to late adulthood after disease burden is significant. The clinical phenotype of MPS I correlates with the specific gene variant involved.
4
In MPS I, loss of function variants are most commonly homozygous or compound heterozygous, and result in significant disruption of gene function with little to no alpha-1-iduronidase activity and an early-onset clinically severe phenotype known as Hurler syndrome.1,2 Patients with Hurler syndrome typically present in infancy with progressive facial coarsening, hepatosplenomegaly, and progressive neurologic deficits. This condition typically causes severe intellectual disability and death in infancy or early childhood if untreated. Patients with more residual alpha-
Several ophthalmologic examination findings have been described in MPS I, including corneal clouding, glaucoma, and pigmentary retinopathy or retinitis pigmentosa.12,13 However, these examination findings are usually documented in childhood due to symptomatic onset early in life. Although retinitis pigmentosa has been previously reported in MPS I, it is an extremely genetically heterogeneous condition, with hundreds of implicated genes, with widely variable inheritance patterns, and can occur as part of systemic syndromes or as an isolated ophthalmologic condition. 14 Due to this genetic and phenotypic heterogeneity, the main commercially available genetic panels for retinitis pigmentosa do not include IDUA, the causative gene for MPS I. Thus, an ophthalmologist is unlikely to make a diagnosis of MPS I-associated retinitis pigmentosa in the absence of other suggestive clinical data and without the assistance of a clinical geneticist.12,13
Once identified, management of MPS I is dependent on disease severity and requires a multidisciplinary approach. Treatment in MPS I consists of palliative/supportive care, early hematopoietic stem cell transplantation (HSCT) with bone marrow or umbilical cord blood stem cells, and ERT with recombinant human α-
The key to preventing irreversible organ damage is effective disease recognition and early diagnosis. Overall, treatment options for lysosomal storage disorders remain limited and so there is a need for innovative therapeutic approaches. Currently, therapeutic options include bone marrow transplant, hematopoietic stem cell transplant, and ERT. Stem cell–based approaches have shown promise in preclinical trials.18 –20 Multipotent adult mesenchymal stem cells (MSCs) derived from bone marrow are of particular interest. MSCs have self-renewing and multi-lineage differentiation capacity. They can be differentiated into mesodermal (osteoblasts, osteocytes chondrocytes, adipocytes, cardiomyocytes), ectodermal (neurocytes), endodermal (hepatocytes) tissues. MSCs are easily accessible and cultured in the clinical setting from the bone marrow of the specific patient (autologous) or from donors (allogeneic). 18 In addition, MSCs are known to be immune-privileged and can be genetically modified with lentiviral or adeno-associated viral transduction to overexpress specific defective lysosomal enzymes. 18 Another novel therapeutic approach employed for MPS I is the autologous transplantation of both hematopoietic stem cells and progenitor cells that have been transduced ex vivo with an IDUA-encoding lentiviral vector after myeloablative conditioning. 20 Preliminary data from these studies show promising results including a similar safety profile to autologous hematopoietic stem cell transplantation, prompt and sustained engraftment of gene-corrected cells and supraphysiologic blood IDUA activity within a month, and decreased urinary GAG excretion. 20
This case is presented to illustrate the diagnostic challenges in recognizing attenuated forms of MPS I; particularly, in individuals born before MPS I was added to the newborn screening panel. In our patient, short stature which may present in the pediatric population as growth failure along with tracheal stenosis, carpal tunnel syndrome, joint contractures, retinitis pigmentosa, and pigmentary retinopathy were the initial red flag signs for attenuated MPS I. By highlighting the factors that contributed to delayed diagnosis and treatment for this patient, we hope to increase awareness across multiple specialties.
Footnotes
Acknowledgements
The authors would like to thank the patient and her family.