
Abstract
Marfan syndrome (MFS) is an autosomal dominantly inherited disorder affecting the cardiovascular, ocular and musculoskeletal systems. Frequently, clinical suspicion and subsequent diagnosis begins in the ophthalmology clinic. Importantly, the ophthalmologist has a responsibility to cater not only to the eye, but also to be involved in a holistic approach for these patients. In this review, we discuss how MFS may present to an eye clinic, including clinical features, ocular morbidity, genetic diagnosis and management. Although this condition is ideally managed by a multidisciplinary team, our focus will be on MFS and the eye, including other conditions which may present with similar phenotypes. The ophthalmologist’s role as the potential first contact for a patient with suspected MFS is crucial in making the proper investigations and referral, with the knowledge that not all ectopia lentis cases are MFS and vice versa. Management of ocular conditions in MFS may range from simple observation to surgical intervention; current options will be discussed.
Plain Language Summary
Eye problems in Marfan Syndrome – A Review
Marfan syndrome (MFS) is an inherited disorder that affects many systems of the body, including the heart, joints, skeleton, skin and eyes. Although the more dangerous problems caused by this are to do with the heart and blood vessels, it is quite often that such patients are first found by eye doctors. They are either seen due to being very short-sighted or with dislocated lenses which can cause major problems in the eye.
Eye problems can be managed by regular observation, although they often require surgery. Because eye doctors are often the first to see these patients, they must involve other doctors of different specialities to help in diagnosing and managing important issues these patients may have, especially affecting the heart and major blood vessels.
Confirmation of diagnosis is done through genetic testing, which has advanced greatly, finding new mutations which may contribute to this disorder. Genetic counselling services can help families in understanding their diagnosis and making better informed decisions about future family planning as well as screening other family members. The eye is just one part of this complex genetic disease. We look in detail at how eye doctors can best approach such patients.
Introduction
Antoine Marfan first described the condition in 1896 that would later be named after him (Marfan syndrome: MFS OMIM 154700). 1 MFS is a multisystem condition, diagnosed according to the revised 2010 Ghent criteria (see Figure 1). 2 Although a relevant family history is considered a positive indicator of the diagnosis, genetic confirmation adds a layer of certainty and is invaluable. It is inherited in an autosomal dominant manner and caused by mutations in the FBN1 gene, which encodes the protein fibrillin-1. Inheritance of the condition is from one affected parent in around 75% of patients. The remaining are thought to be as a result of sporadic mutations. The population incidence is 2–3 per 10,000. 3 Since FBN1 was linked with MFS in 1991, 4 more than 800 mutations in this gene have been identified. 5

Summarised Ghent criteria (2010). A diagnosis of Marfan Syndrome can be made when any ONE criteria is fulfilled. See Appendix 1 for full systemic score.
The major ocular association between ectopia lentis (EL) and MFS is well established. Ocular manifestations are common and best managed with early diagnosis. Frequent associations and their incidence include EL (subluxation or dislocation of the crystalline lens), which has been reported to occur in up to 70% of cases, and rhegmatogenous retinal detachment (RRD) in around one third.6,7 Other manifestations include glaucoma, myopia and corneal flattening. The prevalence of these in MFS is unclear. Herein, we will discuss in detail the genetic architecture, ocular manifestations, diagnostic criteria and management.
Genetics overview
FBN1, a 66-exon gene located on chromosome 15 (15q21), is the major affected gene in MFS. As described in the Ghent criteria, 2 a causative FBN1 mutation with either aortic root dilatation or EL immediately diagnoses MFS and is a crucial criterion in the absence of family history (see Figure 1).
EL causing mutations may lead to post-translational folding defects in the protein. Misfolded fibrillin-1 accumulates in the endoplasmic reticulum, thus leading to haploinsufficiency. There is a predominance of missense mutations affecting cysteine residues in up to 70%; 5 most often resulting in this residue being substituted. 8 The first 15 exons (5′ end) harbour more EL causing mutations, suggesting that the N-terminus of the protein (encoded by this end of the gene) is particularly significant in the aetiology of EL. 9 It has even been suggested that there is an inverse relationship between mutations in the distal part of the gene (3′ end) and EL. 10
Overall, the calcium-binding epidermal growth factor (EGF)-like domains of fibrillin-1 are affected the most. Around 10% of mutations are shared between families. 11
A recent study of over 1500 patients demonstrated 643 different pathogenic variants. 12 The authors broadly categorised their variants into two categories: premature termination codon (PTC) mutations or ‘in-frame’ mutations. Interestingly, they found that EL was less common in those with PTC variants. Rather, they had more severe aortic phenotype with higher risk of aortic dissection. They also confirmed the association with ‘in-frame’ mutations in which cysteine residues were involved.
Another example of genotype–phenotype correlation was demonstrated by Hernándiz et al., 13 in which patients with haploinsufficiency-causing variants had a greater aortic involvement and systemic manifestation. By contrast, dominant-negative mutations, especially including cysteine substitution, more frequently presented with EL.
It has been acknowledged that 25% of disease-causing FBN1 variants are found to be de novo mutations, the remainder unknown. A recent study of 30 families suggested screening for parental somatic mosaicism should be routinely implemented in de novo cases of MFS after they found 2/30 families were found to have somatic mosaicism. It is becoming apparent that parental mosaicism may be more common in MFS than previously thought.14,15 This can clarify clinical signs and symptoms and help in offering appropriate counselling and surveillance. 16
Many autosomal recessive conditions have been demonstrated to cause EL (see Table 1). The most common recessive mutations affect members of the ADAMTS (A Disintegrin and Metalloproteinase with ThromboSpondin) family of proteins, resulting from a loss-of-function mechanism. This family contains 19 enzymatic members, and 7 ADAMTS-like proteins, which lack the protease domain of the ADAMTS proteins. 17
Genes that cause ectopia lentis.
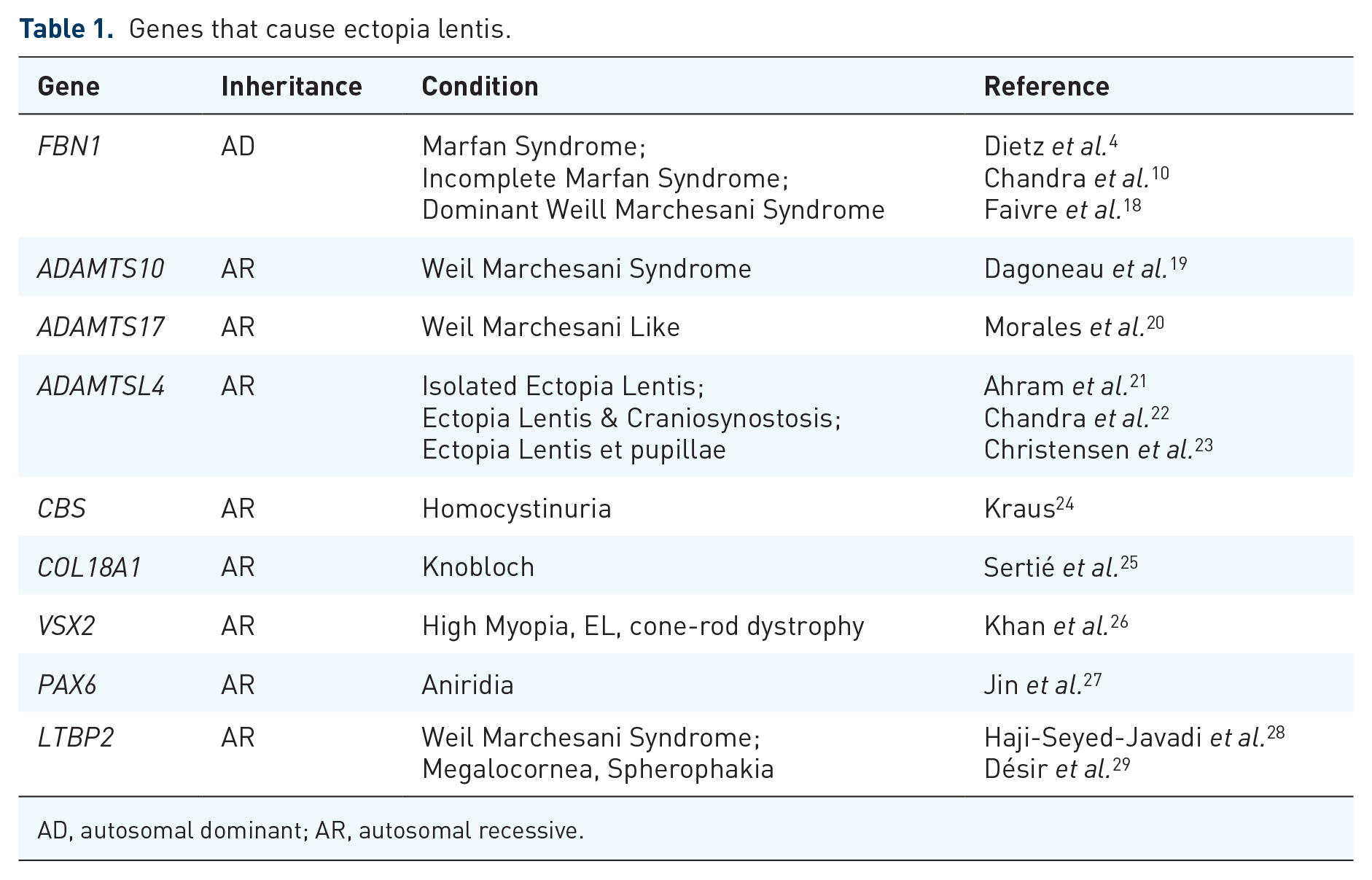
AD, autosomal dominant; AR, autosomal recessive.
The most relevant of these mutations are in ADAMTSL4, located at chromosome 1q21.2. Mutations in this gene cause isolated ectopia lentis (IEL) and Ectopia Lentis et Pupillae (ELP). 22 As the names suggest, these conditions are isolated with no systemic features. The ADAMTSL4 protein has been demonstrated in ocular tissue 30 and has been found to co-localise and co-function with fibrillin-1. 31 Its most relevant role, demonstrated by EL, is likely to be in the structure and function of the ciliary zonule. It has been suggested that ADAMTSL4 has pleiotropic effects, 32 but the gene’s true function is not yet known.
Other ADAMTS mutations have been described (see Table 2) to cause syndromes with EL, including ADAMTS10 (19p13), ADAMTS17 (15q26) and ADAMTS18 (16q23.1).16,33
Recessive mutations in ADAMTS group.

Current genetic testing techniques and analysis
Investigation for a FBN1 mutation is a crucial element of confirming MFS. Relying on clinical criteria alone may lead to false negatives. 42 The Ghent criteria 2 stipulate that identification of a mutation in FBN1, which is recognised to cause MFS, is a major diagnostic criterion.
Most commonly, DNA is extracted from a saliva or blood sample and quantified using fluorescent quantification. Sanger sequencing was traditionally the gold standard method to investigate for mutations; however, it requires the use of MLPA (multiplex ligation-dependent probe amplification) to study possible large deletions. The arrival of next-generation sequencing (NGS) in the past few years has revolutionised genetic interrogation. Many laboratories use genetic panels, 43 while others prefer whole exome sequencing (WES). This investigates the protein coding exons and the intronic/exonic boundaries;44,45 however, the full intronic sequence is not covered. Deep intronic mutations are recognised in MFS, 46 hence whole genome sequencing (WGS) is preferred.
As the name suggests, WGS investigates every nucleotide, including intronic and regulatory regions and structural variants such as copy number variants (CNV) or large deletions, 47 which may be missed with WES. When investigating the whole genome, WGS allows researchers to interrogate mutations outside FBN1, which may cause conditions with very similar phenotypes to MFS. These include ACTA2 in thoracic aortic aneurysm and dissection (TAAD), ADAMTSL4 for IEL or TTLL11 for scoliosis. 48 This is particularly valuable if a variant of unknown significance is found in FBN1.
The data processing from NGS is time-consuming and requires a great knowledge of bioinformatics. The variants identified are analysed and variant interpretation tools are used to further clarify their significance. Finally, causative variants are confirmed with Sanger sequencing in the proband and preferably in affected family members to confirm co-segregation.
There are thought to be over 1000 mutations in FBN1 49 associated with MFS. It is important to interpret any FBN1 variants from the ophthalmic clinic in the context of updated curated databases to clarify if these have been described in MFS. More than 1800 different pathogenic variants in the FBN1 gene have been found, with a large variety of phenotypes. 50 We have previously demonstrated that FBN1 variants causing EL considered insignificant may subsequently become recognised as MFS mutations. 10
The term ‘autosomal dominant isolated ectopia lentis’ has existed for those with FBN1 mutations causing EL without other clinical features of MFS. With this in mind, we showed that 46% of patients described as ‘FBN1 associated autosomal dominant ectopia lentis’ would subsequently be diagnosed as MFS based on their mutation. 10 This suggests that EL caused by novel mutations in FBN1 is actually part of a spectrum of fibrillinopathies with MFS. Thus, in the context of an FBN1 variant with EL, these patients should have regular cardiovascular review. We recommend the term ‘autosomal dominant isolated ectopia lentis’ be dismissed. Instead, we recommend ‘Incomplete Marfan Syndrome’ to highlight the importance of cardiac monitoring. The term ‘isolated ectopia lentis’ should be reserved for recessively inherited EL, particularly if a genetic mutation is found in a recognised gene such as ADAMTSL4. 22
A major study of 1013 probands with MFS showed that 48% did not have a previous family history, either because related carriers were asymptomatic or the mutation appeared sporadically. 5 In the Sonalee laboratory (Imperial College London, UK) in 2021, FBN1 mutation was seen in 789/1186 (67%) participants. These mutations were classified as familial in 446 (56.5%) and sporadic in 343 (43.5%). Further analysis of the 393 probands only found 50 (12.7%) familial variants and 343 (87.3%) sporadic. Participants were classified with a sporadic mutation if there was no other family member in the database with the same mutation. Participants were classified with a familial mutation if there were at least one more family member in the database with the same mutation. Therefore, it is important that even in the absence of a family history, those with suggestive phenotypes are considered for genetic investigation. The advances in investigative techniques have led to mutations being discovered in over 90% of clinically suspicious patients with MFS.
Whether fourth-generation sequencing, a method of sequencing nucleic acids directly in fixed cells used in cancers, 51 comes into use in MFS is yet to be considered. It is of the authors’ opinion that screening cells one at a time is more useful in cancer research due to various somatic mutations in different cells. This also aids in assessing tumour extent.
Although FBN1 mutations have been seen in individual exhibiting mosaicism, this arises when some cells do have the mutation in the heterozygote form and other cells show only the wild type. Fourth-generation sequencing of one cell at a time could be beneficial to confirm the mosaicism in a patient with MFS, but would not have a major role in routine screening with WGS [either with short DNA fragments (Illumina Inc., San Diego, USA) or large DNA fragments (Oxford Nanopore® or PacBio®)]. These large DNA fragment tools are also used mainly for CNV (Copy Number Variation), to interrogate large deletions that otherwise would not have been easily traced with WES or Sanger sequencing. It is clear that the role of genetic analysis in MFS is clearly critical and the future is promising.
Clinical overview and diagnostic criteria
MFS exhibits high inter-familial and intra-familial variability. Its broad phenotypic spectrum ranges from mild symptom free clinical features to rapidly progressive neonatal multisystem disease. 3 MFS typically affects the cardiovascular, skeletal and ocular systems and can also involve the central nervous system, respiratory system and the skin. 2
MFS diagnostic criteria consist of major and minor manifestations in different organ systems (Ghent Criteria). 2 The main clinical manifestations are EL and aortic root aneurysm/dissection. Other clinical features are less influential in the diagnostic evaluation and only contribute to a systemic score (see Appendix 1) that guides diagnosis in the presence of aortic disease but absence of EL. Genetic testing is crucial as it is also the third major criterion. In the absence of relevant family history, the diagnosis of MFS is made when any two of the following three features are present:
A disease-associated variant in the FBN1 gene;
Ectopia lentis;
Aortic root enlargement (Z-score ⩾2.0). 2
The so-called ‘Marfanoid Habitus’ is described as tall, long, slender build 52 associated with a wide range of skeletal findings including arachnodactyly, chest wall deformities and scoliosis. Facial features include a long, narrow face with deep set eyes, down slanting palpebral fissures, flat cheek bones and a small chin. 3
Ocular manifestations
The ocular manifestations of MFS are varied. Visual morbidity is most commonly associated with EL and RRD; these will be discussed in greater detail.
Ectopia lentis
EL is defined as the abnormal location of the crystalline lens as it moves from its natural position. It is described as subluxation if within the pupillary plane or dislocation if the lens moves beyond this into the anterior or posterior segment. Beyond trauma, this condition can be inherited in four major ways:
Isolated Ectopia Lentis (IEL): autosomal recessive inheritance (OMIM # 225100);
Ectopia lentis et pupillae (ELP): autosomal recessive inheritance (OMIM# 225200);
As part of ocular dysgenesis (e.g. aniridia, ADAMTS18 mutations);
As part of systemic syndromes (most commonly MFS: autosomal dominant inheritance OMIM# 154700). 33
The human crystalline lens is held in its natural position behind the iris by the zonular filaments (ZF). Together, they form a ring-like structure between the equatorial lens and the ciliary body. Fibrillins are the major macromolecular component of ZF, the most abundant of which is fibrillin-1. 53 Fibrillin-1 is encoded by FBN1, hence mutations in this gene lead to disruption of the structure and function of fibrillin-1 and zonular dysfunction, causing EL. 54
EL is a hallmark feature (see Figure 2) and is considered an early sign of MFS with most displacements occurring in childhood. However, there is a small risk of developing lens displacement in adulthood. 55

Ectopia lentis in MFS, the edge of the lens can be seen temporally within the pupillary plane.
Lens displacement can range from a subtle posterior tilt (often accompanied by iridodonesis) to frank dislocation. It is typically bilateral and symmetrical although unilateral cases have also been described. 56 Asymmetric zonular deficiency causes the lens to move towards the least affected zonules. Occasionally, only a small notch may be found on careful examination which may be just an incidental finding. If the zonular deficiency is circumferential around the equatorial region of the lens, the result is microspherophakia, which may result in lenticular myopia. If the lens tilts forward, this may cause pupil block, with a subsequent rise in intraocular pressure and accompanying corneal decompensation. Although more common in Weill Marchesani Syndrome, microspherophakia is encountered in other genetic causes of EL. 54
Classifications of EL have been proposed previously. 30 One example is a 5-grade classification by Zech et al., 57 which is for EL specific to MFS. Their defined method includes measurement of visual acuity with and without correction, slit-lamp biomicroscopy, retro illumination and a three-mirror lens. The examination must be done in primary and downward gaze.
Grade 2 is described as that in which there is anteroposterior shift with superior displacement in primary gaze and visualisation of the equatorial part of the lens in downgaze. The authors demonstrated a specificity (100%) and positive predictive value (100%) when using it for clinical diagnosis of MFS. Other clinical features can vary depending on the underlying condition and are determined by the genotype-phenotype. IEL is a condition, as the name suggests, which has no features beyond EL. It is caused by mutations in ADAMTSL4 and most commonly presents in early childhood and tends to have a more severe ocular phenotype than MFS. 22 Other features such as pupillary membranes might point to the diagnosis of ELP (also caused by mutations in ADAMSTL4). 22
Abnormal location of the crystalline lens may be asymptomatic, particularly if mild and not involving the visual axis. In children, particularly EL may be identified incidentally. Symptoms depend on the anatomy, position, clarity and location of the lens. Ectopic lenses are prone to cataract earlier which may be visually significant.
A subluxated lens can be managed conservatively. Optical correction with appropriate glasses or contact lenses (often to correct around the lens itself in the ‘aphakic’ part of the pupil) may be sufficient.
Surgery may be indicated for the following: 58
EL is causing the edge of the crystalline lens to interfere with the visual axis;
Required corrected visual acuity is no longer achievable using optical correction;
Intolerance to aphakic correction;
Troublesome fluctuation of vision caused by the instability of the lens;
To prevent amblyopia;
Anterior dislocation of the lens;
Lens-induced glaucoma or uveitis;
Visually significant cataract.
Surgical removal of the ectopic lens has many options. Patients may initially be left aphakic and managed with contact lenses. This is our preferred initial approach. However, in some cases, intraocular lens (IOL) implantation may be required, either as a primary or secondary procedure. 59
If limited zonular deficiency is found, an anterior approach may be undertaken. The major advantage of this approach would be the possibility of preserving the capsule. Techniques involving Cionni capsular tension rings (CTR) and segments have been reported to have utility in stabilising the capsule for IOL insertion. 60 However, long-term stability has not been reported. It is possible that gradual loss of zonular function may continue, perhaps exacerbated by the surgery. Therefore, our preferred approach is pars plana lensectomy together with vitrectomy which may offer long-term stability. Management of the aphakia in the absence of capsule then offers its own challenges.
FBN1-deficient capsule, zonules, iris and sclera of patients with MFS results in difficult iris and scleral fixation of IOL. Open-loop anterior chamber lenses have been used in MFS with some success, even in paediatric cases. 61 However, the often very deep anterior chambers in MFS may compromise good anterior chamber intraocular lens (ACIOL) fitting resulting in potentially excessive movement of the IOLs. Artisan (iris claw ACIOL) may also provide good results in terms of improving vision, but high incidence of postoperative complications has been reported. 62 Concerns have been raised regarding endothelial cell loss with these IOLs and for this reason surgeons may be reluctant to use such IOLs in younger patients. In an attempt to counter this, retro pupillary iris clip IOLs have been used. 63 Finally, IOLs may be fixed in the sclera in the posterior chamber either with or without sutures. There are numerous techniques available. Comparisons of different techniques are few, with equivalence being demonstrated in some studies. 64
Removal of both the lens and the vitreous gel together reduces the chances of secondary retinal detachment, which has been a significant complication of previous forms of surgery for lens dislocation in MFS. 59 IOL power calculation should be done with caution. Any extreme axial length (AL) can affect the reliability of biometry. 65
Currently, there is no consensus on which type of IOL is most suitable for these patients; therefore, decisions must be made on individual cases. Our preferred technique involves using GORE-TEX® sutures to fix four haptic IOLs. We report recent success with a partially sighted patient diagnosed with EL secondary to MFS. Their visual acuity was reduced to 6/60. With our technique of pars plana vitrectomy (PPV) and four-point fixation IOL with GORE-TEX® sutures, vision improved to 6/9; sufficient for him to hold a driving licence in the United Kingdom (see Figure 3).

Partially sighted individual with MFS: (a) preoperative – ectopic lens with inferior displacement (VA 6/60), (b) intraoperative – Akreos® Insertion, four-point fixation of haptics with GORE-TEX®, Black Arrow shows suture threaded through haptic and (c) postoperative – 6 weeks after surgery, VA 6/9.
Refractive error
Myopia is more common in patients with MFS. The Ghent criteria suggest that myopia of greater than −3DS should be a minor criterion. 2
However, refraction is a complex phenotype with many influencing endophenotypes, including corneal and lenticular structure; both of which are affected in MFS and other EL conditions. Substantial subluxation or dislocation of the crystalline lens may even render the eye hypermetropic and refraction may be somewhat unstable. Therefore, in addition to the relative prevalence of myopia in the general population, we do not feel that refraction serves any diagnostic utility in MFS.
Increased AL is commonly and consistently associated with EL regardless of the genetic aetiology. AL increases particularly when EL is prominent at a young age as a result of form deprivation myopia (FDM) and lens-induced myopia (LIM). 54
Corneas of patients with MFS with EL are flatter and have a higher degree of astigmatism. There is also correlation between eyes with longer AL and flatter corneal measurements. 66 It has even been suggested that corneal curvature may be a useful screening tool for the diagnosis of MFS and shows promising sensitivity and specificity. 67 Although reports regarding central corneal thickness (CCT) are varied, some studies suggest that this is also reduced significantly.68,69
Therefore, we recommend that ocular examination, at least in the absence of EL, also should include full biometric measurements when MFS is suspected. AL should be used rather than refraction as the standard assessment, particularly in children. It has been suggested that AL/Total Corneal Refractive Power ratio may be a potential diagnostic factor for MFS and a better comparison with the myopia of greater than −3D model in terms of specificity and sensitivity. These results indicated that the new model had better discrimination than the myopia of greater than −3D model. 70 Careful examination of the subluxated state of the lens can help clarify the clinical situation and avoid inappropriate prescription.
As with all uncorrected refractive abnormalities, there is the risk of amblyopia. Refractive correction should not be underestimated as a simple and very effective method to manage children with MFS and can be crucial in preventing amblyopia. However, if these measures are insufficient and there is significant progression or once optimal vision is no longer obtainable with refractive correction, early surgical intervention in children with lens subluxation should be considered. 71
Retinal detachment
Posterior segment pathology is reported to be present in 18% of eyes in MFS and the incidence is higher (70%) in patients with a subluxated lens. 72 Retinal detachment (RD) is the most visually significant potential posterior segment complication. Risk factors for RD in MFS include younger age, EL and aphakia. 58 We have previously reported that RD in MFS tends to occur bilaterally (30–42% of cases), and women with MFS seem to develop RD earlier than their male counterparts. 7 This is in contrast to RD affecting those without MFS.
RD can occasionally occur as a complication of surgery to remove a subluxated or dislocated crystalline lens. Although more significant lenticular displacement and high axial myopia are thought to be significant risk factors for RD development after vitreolensectomy, 73 this complication is now much less common with modern surgical approaches. What is unclear is whether the apparent risks of RD are in direct relation to the increased axial myopia in the MFS population or an inherent risk of FBN1 mutations. MFS does not present a clear vitreoretinopathy, clinically. Pathology at the vitreoretinal interface has not been demonstrated.
RD repair can involve either scleral buckling or vitrectomy. The choice of which approach is used will be dependent on the age of the patient, the status of the vitreous, clarity of view and surgeon and patient choice.
Glaucoma
Glaucoma is a common ocular finding in patients with MFS, the most common type being primary open angle glaucoma (POAG); in cases accompanying the syndrome, glaucoma is usually diagnosed at a younger age than in the general population. 74 Microfibril defects could alter elasticity of the trabecular meshwork and episcleral veins, which could impede the pumping action and the pulsatile outflow of aqueous humour, increasing the propensity to ocular hypertension.75–77 Furthermore, alterations to the compliance of the sclera may have an impact on the lamina cribrosa and thus the vulnerability of the ganglion cells. 78
Prostaglandin analogues may be preferred for POAG in MFS. Patients may already be taking β blockers, and it is probably advisable therefore to avoid topical supplementation.
Secondary open angle glaucoma may occur due to RD, vitreoretinal or lens extraction surgeries, iritis, or pigment dispersion due to excessive movement of crystalline or intracapsular IOL.79,80 The latter cause may require peripheral iridotomy. 81
Primary angle closure can result from anterior subluxation of the lens resulting in a pupillary block mechanism. Surgical intervention is therefore often required early in anterior displaced lens. Glaucoma surgery in patients with MFS should be done with caution. Thin sclera and higher risk of hypotony can complicate procedures. 81
Strabismus
Strabismus may present in MFS, and if left uncorrected in children can result in amblyopia. It is suggested to be present in 19% of individuals with MFS, compared to 3–5% in the general population, and may be a presenting sign of the disorder.82,83 Abnormalities in FBN1 in extraocular muscle pulleys and reduced stability may explain the higher incidence of strabismus in patients with MFS. 84
Correction of strabismus is done by extraocular muscle surgery. Since many patients have binocular function potential and are young at diagnosis, if good surgical alignment is achieved, favourable visual outcome may be expected.
Summary
MFS is often first identified in an eye clinic. We have demonstrated its importance and relevance to an ophthalmologist as well as the salient features of the eye. Genetic diagnosis, family counselling and using a multidisciplinary approach are vital.
Once diagnosed, ocular manifestations can be monitored for complications and appropriately managed. We have illustrated the most common conditions. EL is a hallmark feature that must not be underestimated and dealt with in a timely manner. A good visual outcome may be obtained with prompt intervention.
Footnotes
Appendix 1
Systemic manifestations scoring chart.

| Systemic sign | pts |
|---|---|
| Wrist AND thumb sign | 3 |
| Wrist OR Thumb sign | 1 |
| Pectus carinatum deformity | 2 |
| Pectus excavatum or chest asymmetry | 1 |
| Hindfoot deformity | 2 |
| Plain Pes Planus | 1 |
| Pneumothorax | 2 |
| Dural ectasia | 2 |
| Protrusio acetabuli | 2 |
| Reduced Upper Segment/Lower Segment ratio AND increased arm/height AND no severe scoliosis | 1 |
| Scoliosis or thoracolumbar kyphosis | 1 |
| Reduced elbow extension | 1 |
| Facial features (3/5) (dolichocephaly, enophthalmos, down slanting palpebral fissures, malar hypoplasia, retrognathia) | 1 |
| Skin striae | 1 |
| Myopia > 3 diopters | 1 |
| Mitral valve prolapse (all types) | 1 |
Score ⩾7 indicates systemic involvement.
(Maximum total possible: 20 points).
Author contributions
Haseeb Akram: data curation, writing: original draft, writing: review and editing.
Jose Antonio Aragon-Martin: data curation, writing: original draft, writing: review and editing, resources
Aman Chandra: conceptualization, data curation, writing: original draft, writing: review and editing, supervision.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.