
Abstract
Wolfram Syndrome (WS) is an ultra-rare, progressive neurodegenerative disease characterized by early-onset diabetes mellitus and irreversible loss of vision, secondary to optic nerve degeneration. Visual loss in WS is an important cause of registrable blindness in children and young adults and the pathological hallmark is the preferential loss of retinal ganglion cells within the inner retina. In addition to optic atrophy, affected individuals frequently develop variable combinations of neurological, endocrinological, and psychiatric complications. The majority of patients carry recessive mutations in the WFS1 (4p16.1) gene that encodes for a multimeric transmembrane protein, wolframin, embedded within the endoplasmic reticulum (ER). An increasingly recognised subgroup of patients harbor dominant WFS1 mutations that usually cause a milder phenotype, which can be limited to optic atrophy. Wolframin is a ubiquitous protein with high levels of expression in retinal, neuronal, and muscle tissues. It is a multifunctional protein that regulates a host of cellular functions, in particular the dynamic interaction with mitochondria at mitochondria-associated membranes. Wolframin has been implicated in several crucial cellular signaling pathways, including insulin signaling, calcium homeostasis, and the regulation of apoptosis and the ER stress response. There is currently no cure for WS; management remains largely supportive. This review will cover the clinical, genetic, and pathophysiological features of WS, with a specific focus on disease models and the molecular pathways that could serve as potential therapeutic targets. The current landscape of therapeutic options will also be discussed in the context of the latest evidence, including the pipeline for repurposed drugs and gene therapy.
Plain language summary
Wolfram syndrome (WS) is an ultra-rare genetic disease that causes diabetes mellitus and progressive loss of vision from early childhood. Vision is affected in WS because of damage to a specialized type of cells in the retina, known as retinal ganglion cells (RGCs), which converge at the back of the eye to form the optic nerve. The optic nerve is the fast-conducting cable that transmits visual information from the eye to the vision processing centers within the brain. As RGCs are lost, the optic nerve degenerates and it becomes pale in appearance (optic atrophy). Although diabetes mellitus and optic atrophy are the main features of WS, some patients can develop more severe problems because the brain and other organs, such as the kidneys and the bladder, are also affected. The majority of patients with WS carry spelling mistakes (mutations) in the WFS1 gene, which is located on the short arm of chromosome 4 (4p16.1). This gene is highly expressed in the eye and in the brain, and it encodes for a protein located within a compartment of the cell known as the endoplasmic reticulum. For reasons that still remain unclear, WFS1 mutations preferentially affect RGCs, accounting for the prominent visual loss in this genetic disorder. There is currently no effective treatment to halt or slow disease progression and management remains supportive, including the provision of visual aids and occupational rehabilitation. Research into WS has been limited by its relative rarity and the inability to get access to eye and brain tissues from affected patients. However, major advances in our understanding of this disease have been made recently by making use of more accessible cells from patients, such as skin cells (fibroblasts), or animal models, such as mice and zebrafish. This review summarizes the mechanisms by which WFS1 mutations affect cells, impairing their function and eventually leading to their premature loss. The possible treatment strategies to block these pathways are also discussed, with a particular focus on drug repurposing (i.e., using drugs that are already approved for other diseases) and gene therapy (i.e., replacing or repairing the defective WFS1 gene).
Introduction
Wolfram syndrome (WS) is an ultra-rare neurodegenerative disorder defined historically by a cluster of clinical manifestations, namely, diabetes insipidus, diabetes mellitus, optic atrophy, and sensorineural deafness (DIDMOAD).1 –7 A significant proportion of patients will develop additional neurological, psychiatric, endocrine, and urinary tract abnormalities that further complicate the clinical management. Over half of all the patients will develop significant neurological deficits; cognitive impairment is increasingly being recognised as a major neurodegenerative feature. 8 The prevalence of WS has been estimated at 1 in 100,000 in North America and 1 in 770,000 in the United Kingdom.1,9 However, these epidemiological studies predate the discovery of the WFS1 gene (4p16, OMIM 606201) and wider recognition of the more varied clinical manifestations of WS. Using early-onset diabetes mellitus and optic atrophy as the minimum ascertainment criteria for the diagnosis of WS, the estimated prevalence of WS was reported as 1 in 710,000 in Japan. 10 Recent epidemiological studies based on molecular WFS1 typing and a confirmed molecular diagnosis, report an estimated prevalence of 1 in 54,478 in the Messina district of Northeast Sicily, 11 1 in 1,351,000 in Italy, 12 and 1 in 805,000 in North India. 13 Further studies are needed to derive a better estimate of the prevalence of WS in the population, based on a confirmed molecular diagnosis and testing patients who may not exhibit the classic DIDMOAD phenotype. There is currently no treatment that can halt or reverse the neurodegenerative process in WS; management remains largely supportive. 6
Molecular genetics and clinical features of Wolfram syndrome
Wolfram syndrome has been classified into Wolfram syndrome type 1 (WS-1) and type 2 (WS-2) based on the underlying genetic defect and the clinical presentation. Wolfram syndrome type 1 (WS-1), commonly referred to as Wolfram syndrome (WS), is by far the most prevalent type. The majority of patients harbor recessive WFS1 mutations, which encodes for the transmembrane endoplasmic reticulum (ER) protein wolframin.5,14 –16 A subgroup of patients have been reported with dominant WFS1 mutations, which tend to result in a milder phenotype with optic atrophy and sensorineural deafness being particular prominent features.17 –19 Wolframin is a ubiquitous protein that is abundantly expressed in retinal, neuronal, and muscle tissues.14,20 It is a multifunctional protein that regulates a host of cellular functions, in particular the dynamic interaction with mitochondria at mitochondria-associated membranes (MAMs). A more detailed description of the functions mediated by wolframin will be provided in the pathophysiology section of this review.
Although the acronym DIDMOAD is frequently used to describe the classical clinical features of WS, not all patients will exhibit the full phenotype. The development of diabetes mellitus and bilateral optic atrophy before the age of 16 years old is a defining clinical feature of this genetic disorder and, historically, the ‘minimum criteria’ for the clinical diagnosis of Wolfram syndrome.1,3 The pathological hallmark is the degeneration of retinal ganglion cells (RGCs) within the inner retina, resulting in progressive visual loss and optic atrophy, which is diagnosed at an average age of 10–11 years old. 1 In addition to a decline in best-corrected visual acuity (BCVA), there is marked impairment of color vision and a mostly central or caecocentral visual field defect (scotoma). Optical coherence tomography (OCT) imaging reveals diffuse thinning of the retinal nerve fiber layer (RNFL) that is more marked in the temporal quadrant subserving the papillomacular bundle in the early stages of the disease [Figure 1].
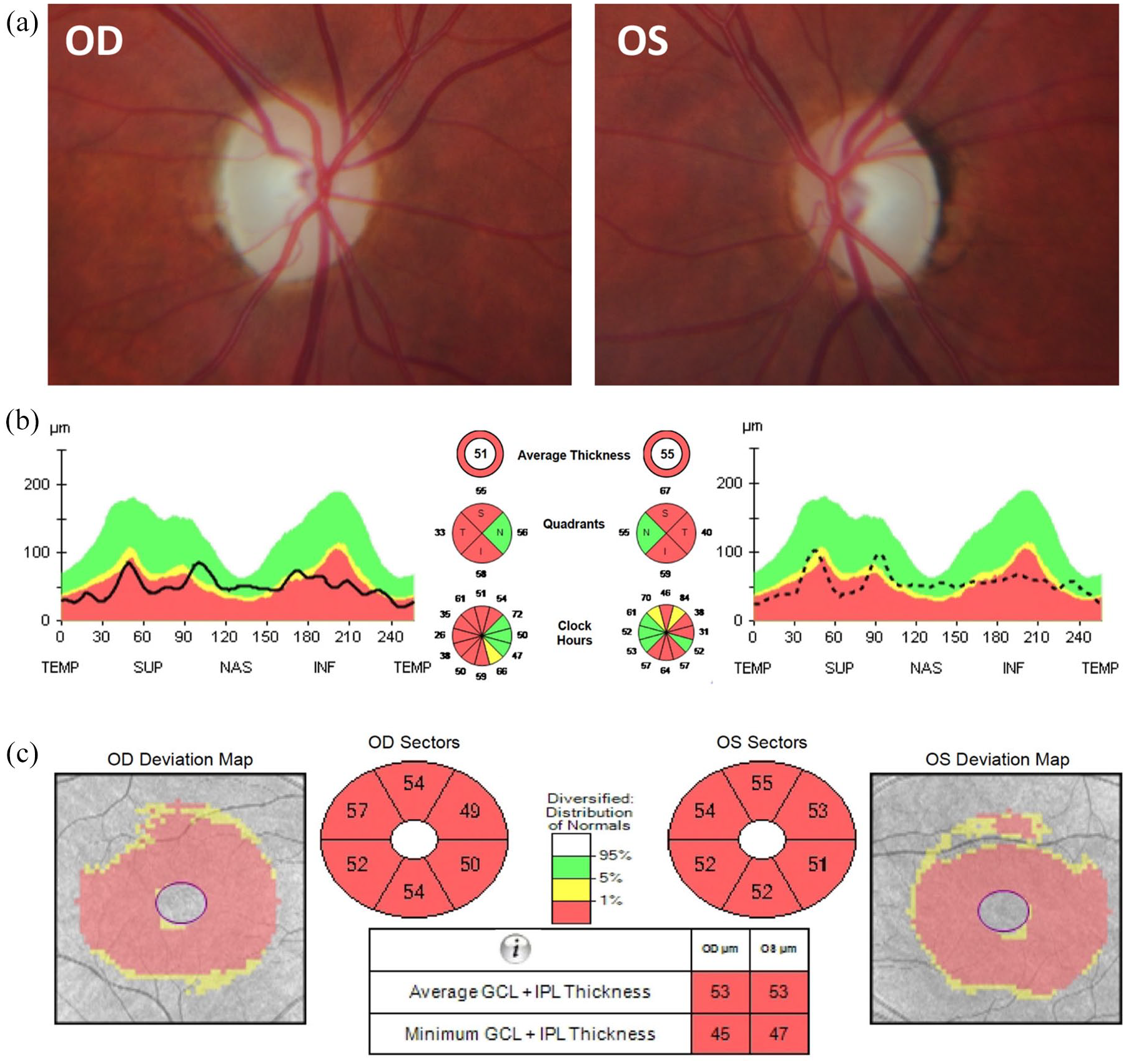
Ophthalmological imaging findings in a patient with WFS1 spectrum disease. A 32-year-old woman developed progressive bilateral visual loss from the age of 27 years. She did not have a history of deafness, diabetes mellitus, or diabetes insipidus. There was no family history of early-onset progressive visual loss. Magnetic resonance imaging revealed small optic nerves without abnormal contrast enhancement. Whole mitochondrial genome sequencing did not detect any pathogenic variants, including the three most common mtDNA mutations associated with LHON. However, she was found to carry compound heterozygous WFS1mutations (c.605A >G p.(Glu202Gly); c.874C >T p.(Pro292Ser)). On examination, the best-corrected visual acuity was 6/24 in both eyes. Color vision was 9/15 Ishihara plates in the right eye (OD) and 7/15 in the left eye (OS). There was no relative afferent pupillary defect. (a) Both optic nerves were pale. (b) OCT imaging of the optic nerves revealed significant retinal nerve fibre layer loss with some sparing of the nasal fibers bilaterally. (c) OCT of the macula showed symmetric generalized atrophy of the GCL and IPL in both eyes.
The development of diabetes mellitus in WS, which is due to the chronic loss of pancreatic β-islet cells, occurs in 98% of WS patients at an average age of 6 years old.2,21 Compared to type 1 diabetes mellitus, patients with WS tend to have better glycemic control. 22 Central diabetes insipidus and sensorineural hearing loss, which are the two remaining features of the full DIDMOAD phenotype, are less common, but still considered major clinical features of WS. 6 Central diabetes insipidus due to loss of vasopressin-producing neurons in the hypothalamus is diagnosed at an average age of 14 years old. 1 Most patients respond well to treatment with intranasal or oral administration of desmopressin. 23 Sensorineural hearing loss emerges in late childhood and its occurrence in WS ranges from 48% to 75%.21,24 Most patients experience slow and progressive hearing loss, with detectable changes having been reported on standard audiological tests for at least two years prior to an individual becoming symptomatic. 24
Although not described in detail by Wolfram and Wagner in their seminal paper, 7 patients with WS also experience significant neurological and urological complications. Neurological abnormalities are found in 53% of patients with WS, 8 with one study reporting a median age of onset of 15 years old and characterized by variable combinations of cerebellar ataxia, peripheral neuropathy, cognitive impairment, dysarthria, epilepsy, and autonomic dysfunction.6,8,23 Urinary tract problems, including dilation of the urinary tracts, neurogenic bladder, and urinary incontinence, have been reported in 19% of patients with WS, but this figure is likely to represent an underestimate.23,25 There is evidence that lower pontine volume on magnetic resonance imaging (MRI) is associated with more severe urinary tract dysfunction and lower quality of life in WS. 25
With greater access to genetic testing and the application of next-generation sequencing in clinical practice, there is increasing recognition of a subgroup of patients who harbor heterozygous dominant pathogenic variants in the WFS1 gene.17 –19 These are more often missense pathogenic variants, compared to the loss-of-function variants and deletions identified in patients with recessive WFS1 mutations. 26 Although the phenotypic spectrum of patients with dominant variants in the WFS1 gene has not yet been fully defined in a large cohort of patients, the disease is usually less severe from a neurological perspective. There is no universally accepted nomenclature; the terms Wolfram syndrome-like disorder, Wolfram syndrome spectrum disorder, and WFS1 spectrum disorder have all been used to define the more limited phenotype seen in patients with not only dominant, but also recessive, WFS1 mutations. 27 Optic atrophy, with or without sensorineural hearing loss, is a frequent manifestation of the dominant form of the disease. 18 In rare instances, congenital cataracts have been described in families segregating dominant WFS1 mutations. 28
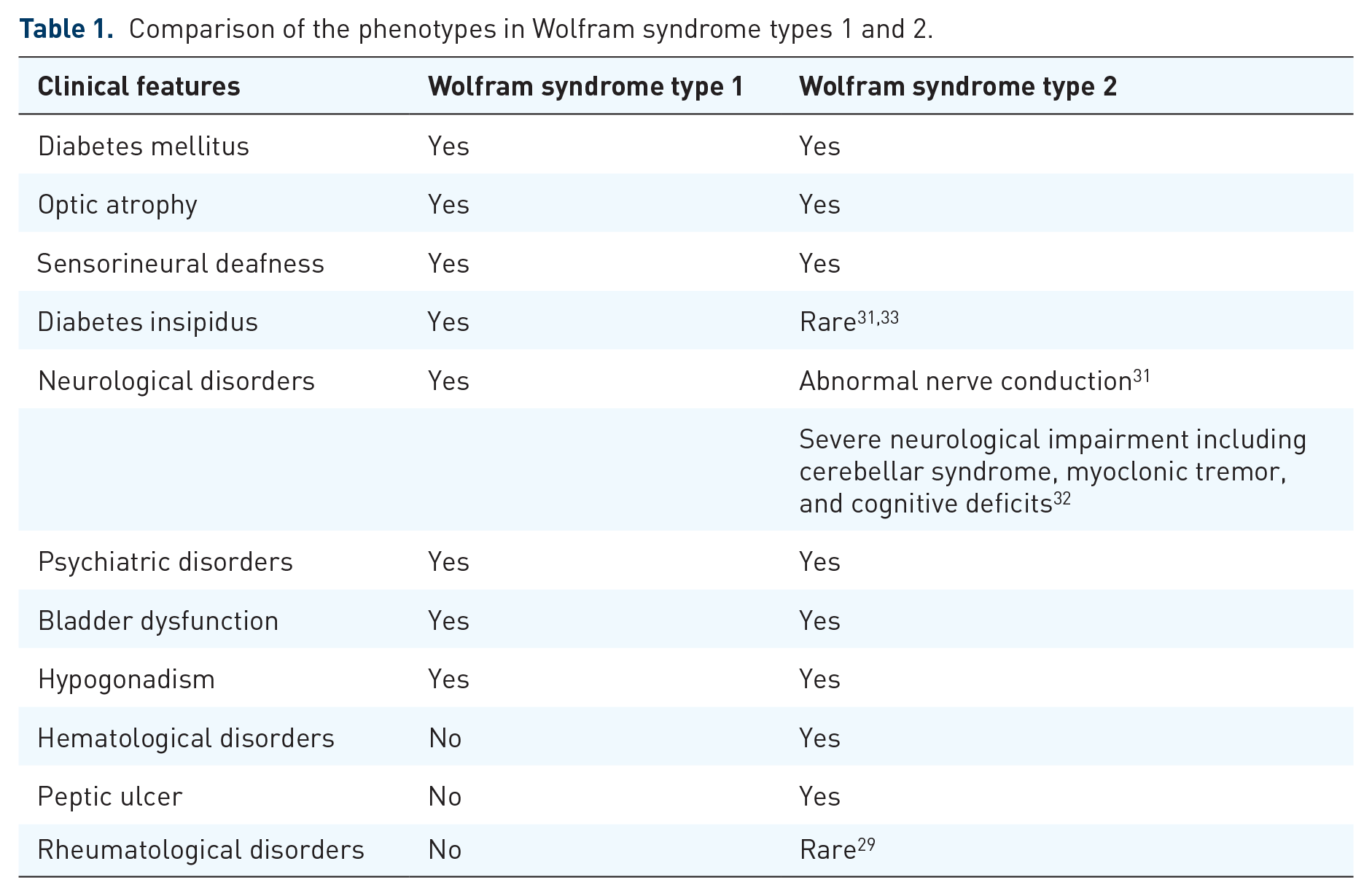
Only a few patients with WS-2 have been reported worldwide carrying recessive mutations in CISD2 (4q24, OMIM, 604928), which encodes for the ER intermembrane small protein CDGSH-sulfur domain-containing protein 2 (CISD2). 29 Patients with WS-2 share some of the clinical features seen in WS, including the development of diabetes mellitus, optic atrophy, and sensorineural hearing loss in the first two decades of life.29 –32 However, WS-2 is typically associated with peptic ulcer disease and bleeding tendencies without diabetes insipidus (Table 1). 29
Comparison of the phenotypes in Wolfram syndrome types 1 and 2.
It is clear that we need to move towards a classification of disease based on the molecular confirmation of the underlying pathogenic mutation (WFS1 or CISD2) and the mode of inheritance (dominant or recessive).19,34,35 DIDMOAD should be viewed as a historical description from the premolecular era and Wolfram syndrome is best considered as a spectrum disorder with major and minor clinical features that can occur in various combinations (Table 2). The presence of any combination of these features should prompt further investigations, including confirmatory genetic testing, even if the ‘minimum criteria’ of diabetes mellitus or optic atrophy are absent.
Clinical manifestations of Wolfram syndrome.
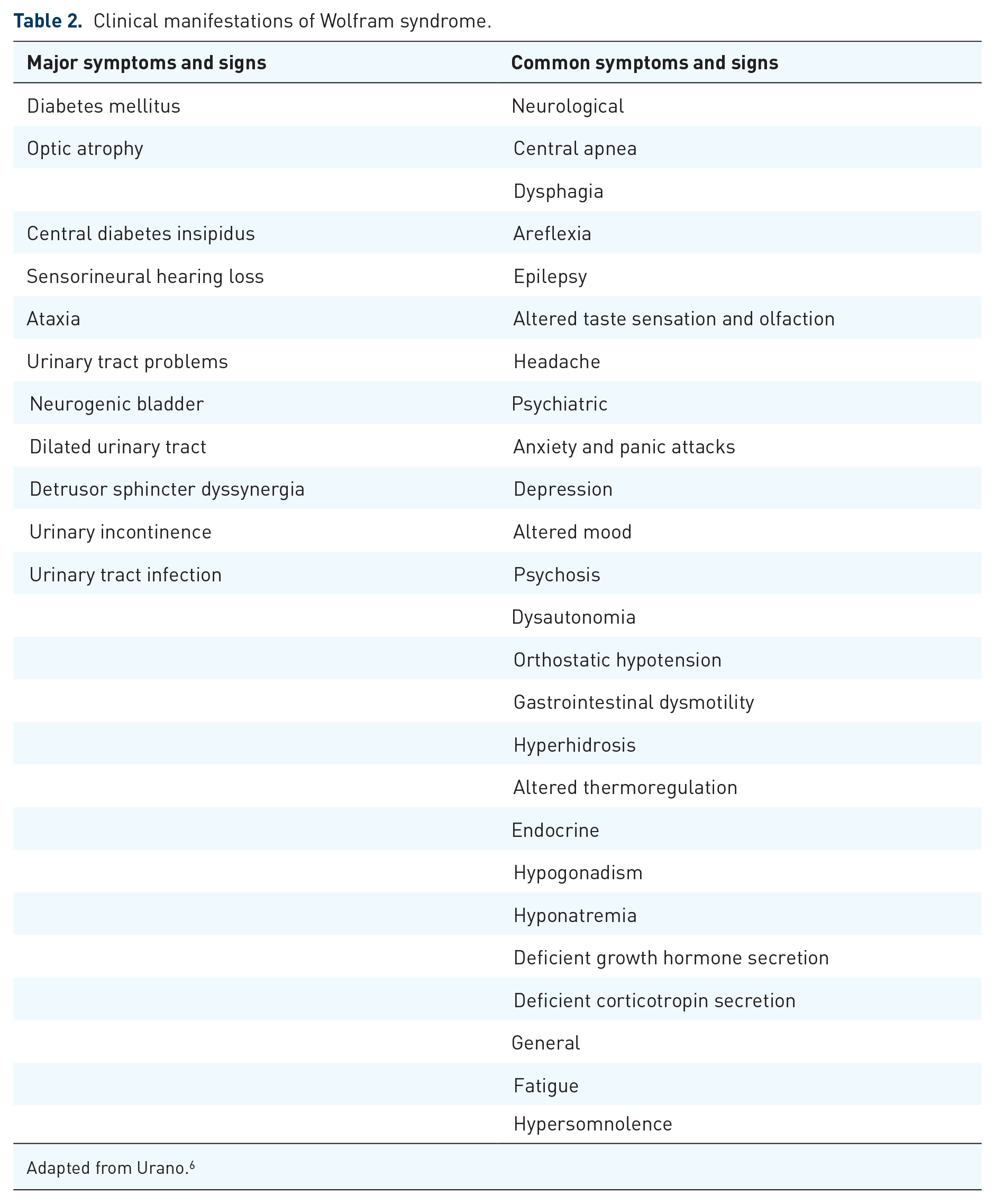
Adapted from Urano. 6
Selective vulnerability of retinal ganglion cells in Wolfram syndrome
Wolframin is highly expressed in the retina, including the photoreceptor inner segments, the inner nuclear layer (INL), and RGCs. 36 For reasons that are still unclear, RGCs within the inner retina are preferentially vulnerable to the deleterious consequences of WFS1 mutations. The progressive loss of RGCs and their projecting axons results in optic atrophy and irreversible visual failure, usually with central or caecocentral scotomas. This pattern of RGC loss is shared with two other mitochondrial optic neuropathies: Leber hereditary optic neuropathy (LHON), which is caused by mitochondrial DNA (mtDNA) point mutations, and dominant optic atrophy (DOA), which is commonly associated with mutations in OPA1, a nuclear gene that regulates mitochondrial morphology and biogenesis. 37 However, the tempo of visual failure differs between the three conditions. Vision loss in LHON is subacute, usually occurring in the second and third decade of life, and begins as a unilateral, progressive optic neuropathy with sequential involvement of the fellow eye months to years later. 37 In contrast, vision loss in DOA typically occurs in the first or second decade of life, is commonly bilateral, and is relatively symmetric with a mild insidious course. 37 Vision loss in WS starts in early childhood and, generally, the rate of visual deterioration is faster compared with OPA1-related DOA.
Peripapillary and macular OCT is widely used in clinical practice to detect and quantify RGC loss (Figure 1). There is evidence that macular OCT could prove useful in differentiating between patients with dominant and recessive WFS1 mutations. In one study, patients with dominant WFS1 mutations exhibited a distinct lamination defect of the outer plexiform layer (OPL), with abnormal reflectivity that was not observed in WS patients with recessive mutations. 18 The aetiology for this OCT finding is uncertain, but it could possibly be due to the specific effect of dominant WFS1 mutations on Müller cells, leading to the development of OPL and INL oedema and changes within Henle’s fibre layer.
The histopathological pattern of optic nerve atrophy in WS is strikingly similar with that of LHON and DOA. 38 Despite differences in the underlying genetic defects that give rise to WS and LHON, all three conditions exhibit RGC loss in the macula corresponding to the papillomacular bundle. Recent studies utilizing OCT angiography (OCTA) to non-invasively characterize vascular pathology in WS showed similar changes to those observed in LHON. Namely, these were attenuation of the superficial capillary plexus of the temporal optic nerve head and peripapillary microvasculature, as well as peripapillary telangiectatic blood vessels and vascular tortuosity. 39 Although further investigations are required, OCTA could prove a useful biomarker for the inherited optic neuropathies.
Disease models of Wolfram syndrome
Wolfram syndrome type 1 models
Several in vivo and in vitro models of WS-1 have been developed to study the molecular mechanisms underpinning the pathophysiology of WS. Fibroblasts derived from patients carrying recessive WFS1 mutations showed disturbed mitochondrial function and impaired calcium homeostasis, with increased Ca2+ flux from the ER to the mitochondria. 32 In a study with primary cortical neurons from Wfs1-deficient mice, the loss of Wfs1 induced ER stress, resulting in inositol 1,4,5-triphosphate receptor (IP3R) dysfunction and disturbed calcium homeostasis; in addition, there was also evidence of altered mitochondrial dynamics. 40 Inhibition of mitochondrial fusion, alteration in mitochondrial trafficking, and augmented mitophagy were observed in the Wfs1-deficient neurons, which contributed to delayed neuronal development. 40
In a study with Drosophila melanogaster, the fly homolog of WFS1 (wfs1) was knocked down in the central nervous system and the wfs1-deficient flies showed age-related locomotory behavior defects starting prematurely at the age of 21 days. 41 The wfs1-deficient flies were more prone to oxidative stress, as exposure to hydrogen peroxide increased the levels of wfs1 mRNA in the fly brain. 41 However, ER stress or defects in mitochondrial dynamics were not observed in the wfs1-deficient flies. 41 Deficiency of wfs1 in both neurons and glial cells further exaggerated the neurodegeneration, behavioural deficits, and premature death observed in the flies. 41 Interestingly, the expression of wfs1 RNA increased with ageing in the fly brain, suggesting a possible role in the development of age-related neuronal disorders in addition to causing monogenic WS. 41
Different vertebrate models have been developed, including transgenic rat, mouse, and zebrafish models, to study the molecular functions of WFS1 and the factors contributing to disease progression in WS. A mutant rat line created by deletion of exon 5 demonstrated a reduction in pancreatic β-cell mass and increased pancreatic ER stress markers. 42 The retinal and brainstem expression of ER stress markers were also increased, with a reduction in optic nerve and medullary volumes. 42
Characterization of Wfs1 expression in the mouse central nervous system revealed that Wfs1 is enriched in the central extended amygdala, the CA1 region of the hippocampus, and the nucleus accumbens, suggesting a potential role of Wfs1 in regulating anxiety and the stress response. 43 In another study, Wfs1 and its related molecules have been proposed as key molecules in the hippocampus associated with the early- and long-term development of depression, although the detailed functions of these molecules in the pathological progression of depression remain poorly understood. 44 Behavioral studies of Wfs1 mutant mice show altered responses when they are exposed to induced stressful environments, for example, decreased locomotor response in abrasive brightly lit conditions.45,46 A subpopulation of Wfs1 mutant mice produced a spontaneous audible chirping sound that increased in loudness under stressful situation, and these were suppressed by the administration of the anxiolytic drug diazepam. 45 In another study, Wfs1 mutant mice showed freezing behavior, decreased social interaction, and reduced learning and memory, in keeping with the neuropsychiatric features observed in the patients with WS. 46
Zebrafish models of WS have recently been characterized in our laboratory with mutations in wfs1a and wfs1b. Zebrafish have two orthologs of the WFS1 gene due to an evolutionary duplication event. The wfs1b-/- mutant zebrafish better recapitulated the pathophysiological hallmarks of human WS with progressive RGC loss and reduced visual function. 47 Furthermore, wfs1b-/- zebrafish embryos showed specific neural circuit abnormalities that likely contributed to the locomotor defects observed in the mutant larvae and adult fish. This particular zebrafish model provides a powerful tool, not only to carefully dissect the disease mechanisms in WS, but also as a versatile system for therapeutic drug screening.
Wolfram syndrome type 2 models
Relatively few studies that have explored how the loss of CISD2 function leads to human disease. In neuronal cells, CISD2 is thought to be directly involved in the regulation of intracellular calcium levels and apoptosis. 48 In patients with hepatocellular carcinoma, higher levels of CISD2 expression were associated with a shorter life expectancy and a higher rate of tumour recurrence. These results were confirmed in vitro, with the downregulation of CISD2 expression in hepatocellular carcinoma cells suppressing cell proliferation. 49
There are few in vivo studies about the behavioural and physiological roles of CISD2 in WS-2. In a particular Cisd2 murine model, mutant mice exhibited degeneration of skeletal muscles, upregulated autophagy, dysregulation of calcium homeostasis, and morphologically enlarged mitochondria. 50 CISD2 forms a complex with BCL2, which regulates Ca2+ in apoptotic cells and maintains the basal level of autophagy in skeletal muscles. 50 Cisd2 mutant mice also showed features of mitochondrial dysfunction, an increase in autophagy, and severe muscle and neuronal cell degeneration causing premature ageing and a shortened life span. 51 In a behavioural study, Cisd2 mutant mice showed similar type of vocal chirping sound as observed in Wfs1 mutant mice, pointing towards some pathophysiological similarities,45,52 The Cisd2 mutant mice showed decreased body size and weight; they were also hypoactive in behavioural assays, with reduced locomotor activity, impaired spatial learning and memory, and exhibiting abnormal stress responses compared to their wild-type litter mates. 52 These Cisd2 mutant mouse models could, therefore, prove useful in further studying the disease pathways precipitating disease progression in patients of WS-2.
Unravelling the pathophysiology of Wolfram syndrome
Wolframin regulates a host of cellular functions, including the dynamic interaction with mitochondria at MAMs. It is a multifunctional protein that has been implicated in several crucial cellular signaling pathways, including insulin signaling, calcium homeostasis, and the regulation of apoptosis and the ER stress response (Figure 2).20,53 –55

Pathophysiological mechanisms implicated in Wolfram Syndrome. (a) A mutation in the WFS1 gene can lead to loss of function (with degradation of the mutant mRNA transcript) or result in an aberrant wolframin protein with a dominant-negative effect or a gain-of-function. In this diagrammatic representation, the mutant mRNA transcript generates a misfolded protein that is mislocalised, leading to increased levels of ER stress. (b) Sodium valproate is a drug targeting ER stress that has been shown to increase the expression of p21, which is crucial for cell survival under conditions of heightened ER stress. In healthy cells, wolframin negatively regulates ATF6α, which is a key protein regulating the UPR. Deficiency of wolframin is thought to increase ATF6α signaling, resulting in upregulation of the UPR and increased apoptosis. (c) Wolframin plays a central role in maintaining ER homeostasis and in controlling intracellular Ca2+ flux. Loss of wolframin increases cytosolic Ca2+ and this ultimately triggers apoptosis. The disruption of Ca2+ flux leads to hyperactivation of calpains, which are calcium-dependent cysteine proteases, in particular calpain 2. As a result, there is increased caspase-3 cleavage. CISD2 is a negative regulator of calpain 2 that limits the cleavage of caspase-3. ER Ca2+ stabilizers can normalize ER homeostasis by targeting ER calcium transporters such as the ryanodine receptors. Dantrolene sodium and calpain inhibitors normalize cytosolic Ca2+ and calpain activity by inhibiting ER Ca2+ efflux via the ryanodine receptors, which helps to suppress apoptosis. (d) Wolframin activates IP3R-mediated Ca2+ release and it promotes ER-mitochondria Ca2+ transfer by binding to the NCS1 to form a complex with IP3R. Loss of wolframin triggers NCS1 degradation and IP3R dysfunction leading to decreased mitochondrial Ca2+ uptake, causing mitochondrial dysfunction and a reduction in ATP production. The physical interaction of mitochondria with the ER is established through MAMs. MAMs serve as close contact sites facilitating Ca2+ transfer via IP3R on the ER, which interacts with VDAC1 on the outer mitochondrial membrane. Wolframin deficiency results in disorganization of the MAMs and a reduction in mitochondrial Ca2+ uptake. Ibudilast restores the resting cytosolic Ca2+ levels through its interaction with NCS1.
Endoplasmic reticulum stress and cellular apoptosis
Wolframin is key to maintaining ER homeostasis and WS is a paradigm for ER stress-driven diseases. The ER is the largest organelle in the cell, playing a pivotal role in protein synthesis, folding, modification, and transport. It is also a major store of intracellular Ca2+ ions. When homeostasis is disrupted, unfolded/misfolded proteins accumulate and a state of ER stress ensues, with activation of a network of signaling pathways to mitigate stress and generate proteins for survival, known as the unfolded protein response (UPR). Activation of the UPR results in upregulation of gene expression for molecular chaperones, expansion of the size of the ER, a reduction in protein translation, and degradation of abnormal proteins that have accumulated in the ER. The UPR is beneficial, but it is unable to reduce stress or reset homeostasis under conditions of chronically elevated stress, which leads to a maladaptive response and an enhanced susceptibility to cellular apoptosis. 56 There is evidence that WFS1 and CISD2 mutations may also disrupt cellular Ca2+ homeostasis, exerting a deleterious influence on ER-mitochondrial structure and function by disrupting MAMs. 57
Wolframin controls a regulatory feedback loop of the ER stress signaling network by forming an ER stress-mediated complex with activating transcription factor 6α (ATF6α), which is one of the three UPR activating proteins. 53 Using rodent and human β-cell lines, one study found that wolframin negatively regulates ATF6α through the ubiquitin-proteasome pathway. 53 Wolframin suppresses ER stress signaling by stabilizing E3 ubiquitin ligase (HRD1) and enhancing ubiquitination and proteasome-mediated degradation of ATF6α. Wfs1 knock-out mice had undetectable HRD1 expression in the pancreatic islets and HRD1 expression was also reduced in fibroblasts derived from WS patients when compared with controls. Under conditions of ER stress, ATF6α was released from HRD1 by dithiothreitol and thapsigargin, the latter being an inhibitor of sarco-endoplasmic reticulum Ca2+ATPase. Overexpression of HRD1 enhanced ATF6α protein degradation and ubiquitination. 53
Wolframin plays a central role in the regulation of insulin signaling and pancreatic β-cells. This was recently investigated using a Wfs1-knockout mouse model and tetracycline-inducible β-cells. 20 The knockout mice showed a progressive decline in β-cell mass, disrupted islet architecture, and considerably less total pancreatic insulin content compared with wild-type mice. 20 When wolframin-overexpressing β-cells were placed under chemical ER stress, there was suppression of pro-apoptotic pathways downstream of ATF6α and upregulation of insulin biosynthesis, including upregulation of genes defining β-cell maturity. 20 As a result, wolframin appears to mitigate apoptosis by downregulating the CHOP-mediated apoptotic pathway (CHOP-TRIB3 axis), thereby promoting the activation of the Akt pathway for cell survival.
In another study, Wfs1 mutant mice demonstrated glucose intolerance and diminished insulin secretion in pancreatic β-cells. 58 The isolated islets from the mutant mice indicated loss of β-cells, impaired glucose homeostasis and increased apoptosis, similar to the pathological features observed in patients with WS. 58 Interestingly, the severity of the diabetes caused by Wfs1 disruption was dependent on the genetic background of the mice, that is, more than 60% of the F2 mutant mice [(129Sv × B6) × B6] developed overt diabetes whereas mutant mice on the B6 background only had impaired glucose tolerance, but not overt diabetes. 58 The effects of the mouse genetic background on glucose homeostasis have also been reported previously. 59
CISD2 has been found to negatively regulate calpain 2, a calcium-dependent protease. Calpain 2 is a heterodimer, consisting of the CAPN2 catalytic subunit and CAPNS1 regulatory subunit. Overexpression of CAPN2 in HEK293 cells led to an increase in caspase-3, the latter being a significant regulator of cellular apoptosis. 48 In mouse neuronal cells with suppression of CISD2 expression and in CISD2-knocked down rodent neuronal cells, an increase in the cleavage of caspase-3 was observed under normal and ER-stress conditions. Calpain 2-associated apoptosis was significantly suppressed by ectopic expression of CISD2 and partially suppressed by silencing CAPN2 in CISD2-deficient cells. Treatment with calpeptin, a calpain inhibitor, prevented CISD2-knockdown-mediated cell death in neuronal and β-cells. These studies suggest that CISD2 is a negative regulator of calpain 2-mediated apoptosis. The link between wolframin loss of function and calpain activation was studied using brain tissue from Wfs1 brain-specific knockout and control mice. 48 A significant increase in calpain-specific spectrin cleavage product and myelin basic protein, which is a known substrate of calpain in brain tissue, was observed in Wfs1 knockout mice. Neural progenitor cells derived from the induced pluripotent stem cells (iPSCs) of WS patients were also observed to have increased calpain activity and spectrin cleavage product. As a result, both WFS1 and CISD2 play a substantial role in the regulation of ER stress and apoptosis, and targeting these pathways are promising therapeutic avenues for WS. 48
Impaired calcium homeostasis
The role of wolframin in the regulation of ER Ca2+ levels was first demonstrated in 2003. 60 The overexpression of wild-type wolframin in Xenopus oocytes increased cytosolic Ca2+, indicating an increased release of calcium from the ER into the cytoplasm. 60 The application of IP3 to microsomal membranes from heterologous wolframin-expressing oocytes was followed by an increased slope of conductance. Wolframin was found to regulate ER Ca2+ levels by increasing the rate of Ca2+ uptake into the ER.60,61 The evidence accumulated to date suggest that wolframin is operating as a novel ER calcium channel or as a regulator of ER calcium channel activity.
Another study demonstrated that wolframin deficiency affects the functioning of the IP3R, leading to altered cytosolic Ca2+ homeostasis. 40 In rat neuronal cells, lack of wolframin reduced the amplitude of IP3R-mediated Ca2+ release compared with controls, leading to higher cytosolic Ca2+ in resting conditions. The effects of wolframin deficiency phenocopied the effect of pharmacological inhibition of IP3R by Araguspongin B. Overexpression of the active IP3R fragment restored IP3R-mediated Ca2+ release. Mitochondrial dynamics was affected in Wfs1-deficient neurons, with impaired mitochondrial movement and mitophagy, and an imbalance between mitochondrial fusion and fission. These disturbances were corrected by pharmacologically activating L-type Ca2+ channels. 40 As a result, lower ER Ca2+ release seems to impair mitochondrial dynamics in the context of neuronal development.
Fibroblasts carrying pathogenic WFS1 mutations derived from WS patients demonstrated a reduction in the amount of Ca2+ released from the ER compared with controls. 62 Stimulation with bradykinin, which induces ER Ca2+ release through IP3 receptors, resulted in lower Ca2+ accumulation in mitochondria and the cytosol. Subcellular fractionation revealed that wolframin was present in considerable amounts in the MAMs compartment, highlighting the importance of wolframin in the regulation of Ca2+ at MAM sites. In contrast to other studies, alteration in Ca2+ dynamics did not result in mitochondrial Ca2+ accumulation. 62
In another study using patient-derived fibroblasts, wolframin formed a complex with neuronal calcium sensor 1 (NCS1) and IP3R to promote calcium transfer between the ER and mitochondria. 54 In wolframin-deficient fibroblasts, there was an almost 50% reduction in NCS1 protein levels. In NCS1-knocked down control fibroblasts, there was also a reduction in IP3R-mediated Ca2+ uptake by the mitochondria and in the number of MAM contact sites. NCS1-depleted cells displayed a decrease in mitochondrial complex I- and complex II-driven respiration, similar to fibroblasts of WS patients. 57 Overexpression of NCS1 in wolframin-knockout rat insulinoma cells rescued both ATP-evoked ER calcium release and the resting cytosolic calcium. 63 In mutant patient-derived fibroblasts, overexpression of NCS1 restored ER-mitochondria interactions and Ca2+ exchange, with an increase in complex I- and complex II-driven respiration. 54
Mitochondrial dysfunction
Wolframin deficiency impairs mitochondrial dynamics and this feature is associated with delayed neuronal development and impaired neuronal survival.32,40 Wolframin deficiency induces ER stress leading to IP3R dysfunction, disturbed Ca2+ homeostasis, and altered mitochondrial dynamics. 40 Silencing of the WFS1 gene in HEK cells with small interfering RNA (siRNA) upregulated genes related to mitochondrial dysfunction and degeneration, suggesting that loss of wolframin precipitates degeneration via mitochondrial dysfunction. 51 In rat neurons treated with Wfs1 small hairpin RNA (shRNA), a significant three-fold decrease in the number of mitochondrial fusion events and a decrease in the mitochondrial fission rate were observed. 40 These changes in the fusion-fission cycle led a 20% decrease in mitochondrial length. Studies of mitophagy with LC3 and Keima assays demonstrated that wolframin deficiency increases mitochondrial removal by mitophagy, resulting in fewer mitochondria in axons. Furthermore, wolframin-deficient neurons have significantly reduced axon length during early stages of development. 40 Although relatively mature neurons had similar axonal length and branching to controls, there was a significant reduction in the density of synapses and impaired survival, highlighting the role of wolframin in maintaining normal mitochondrial function and axonal integrity.
Similar to WFS1, CISD2 also plays a crucial role in regulating Ca2+ dynamics and mitochondrial function.30,32,51 Patient-derived fibroblasts carrying a homozygous CISD2 mutation showed a number of mitochondrial abnormalities, including increased ER-mitochondrial contact and a hyperfused mitochondrial network. 32 However, a direct interaction between WFS1 and CISD2 has, so far, not been established.25,32,50
Therapeutic strategies for Wolfram syndrome
The current management of WS is supportive due to the lack of disease modifying treatments that can halt the progressive neurodegeneration in this disorder. Despite many challenges, the advances made in our understanding of the underlying disease mechanisms have led to the development of several therapeutic approaches that are either in preclinical development or at the stage of clinical trials. The strategies being considered include chemical chaperones to optimize the structure of mutant WFS1 proteins, ER calcium stabilizers that target ER calcium transporters, molecules that regulate ER-stress mediated cellular apoptosis and activate cell survival mechanisms, modulators of mitochondrial function, and gene replacement therapy and gene editing to correct a pathogenic WFS1 variant (Figure 3).

Wolfram Syndrome treatment pipeline. Summary of current therapeutic strategies for the treatment of Wolfram syndrome at different stages of preclinical and clinical development. This figure was adapted with permission from Dr. Sarah Gladstone and the Wolfram Syndrome Research Alliance (WRSA) (https://www.wsresearchalliance.org/treatment-pipeline.html, accessed on the 27th of June 2021).
ER calcium stabilizers
Dysregulation of ER Ca2+ homeostasis is a major pathophysiological player in WS, which leads to upregulation of cellular apoptosis. Stabilizing ER Ca2+ homeostasis, by targeting the pathway leading to calpain activation and calpain-mediated cellular apoptosis, provides an obvious therapeutic target for WS. A recent study using wolframin-knockout insulinoma cells showed that pharmacologic intervention with calpain inhibitor XI, a reversible inhibitor of calpain 1 and 2, normalized resting cytosolic calcium and rescued the viability of cells back to baseline. 63 Wolframin-knockout insulinoma cells had a significantly lower rate of glucose-stimulated insulin secretion compared with wild-type cells; treatment with calpain inhibitor XI reversed this impairment and rescued insulin secretion. Similar findings were observed in cells treated with ibudilast, an inhibitor of cyclic nucleotide phosphodiesterase (PDE), in particular PDE4. Ibudilast is hypothesized to normalize calcium through its interactions with NCS1. In vitro studies have highlighted the importance of NCS1 in regulating calcium homeostasis and cell survival57,63 and its potential as a drug target for WS. 64
Calpain activation is tightly regulated by cytosolic calcium levels. There is interest in finding small molecule compounds capable of altering cellular calcium levels and preventing calpain activation in WS patients. Dantrolene, which is an inhibitor of ryanodine receptors in the ER and a suppressor of calcium efflux from the ER to the cytosol, is under investigation in a phase Ib/IIa non-randomised, open-label, clinical trial [ClinicalTrials.gov identifier: NCT02829268]. Dantrolene treatment in rat insulinoma cells and mouse neural cells decreased cytoplasmic calcium levels and restored cytosolic calcium levels in wolframin-knockdown cells. 48 Suppression of apoptosis and calpain activity were also observed in these cells. These findings were replicated in pre-treated neural progenitor cells derived from iPSCs of a patient with WS carrying recessive WFS1 mutations. 48
Modulating ER stress
Suppression of ER stress-induced apoptosis and activation of pathways that promote cell survival after ER stress have been the focus of several studies. Sodium valproate, which is used in the treatment of epilepsy and bipolar disorder, is currently under investigation. Although further work is required to precisely define its molecular target(s), sodium valproate is thought to increase the level of p21, a cyclin-dependent kinase inhibitor that exerts a protective function against cellular stress, regulates the cell cycle, and importantly, exerts an anti-apoptotic effect.65,66 In cells exposed to thapsigargin, a naturally occurring plant extract that causes apoptosis in cells via activation of ER stress, administration of sodium valproate improved cell viability with an increase in p-AKT level and inhibition of ER-stress induced apoptosis response proteins, including CHOP. 67 Sodium valproate also modulated ER stress in neuronal cells by inducing expression of the WFS1 protein and regulating glucose-regulated protein 94 (GRP94), an essential ER chaperone protein. 68 In HEK cells carrying dominant WFS1 mutations, the levels of ER stress and CHOP were reduced following treatment with sodium valproate or 4-phenylbutyric acid (PBA), a low molecular weight fatty acid and chemical chaperone. 69 By stabilizing protein conformation during folding, ameliorating trafficking of mutant proteins, and suppressing unfolded protein aggregation, another study found that PBA restored normal insulin synthesis and the ability to upregulate insulin secretion in WFS1-deficient β-cells differentiated from patient-derived iPSCs. 70 The efficacy of sodium valproate in delaying neurodegeneration and visual deterioration is currently being evaluated in a phase III randomised, double-blind, placebo-controlled trial [ClinicalTrials.gov identifier: NCT03717909]. There are currently no clinical trials of PBA in WS patients.
Glucagon-like peptide-1 receptor (GLP-1R) agonists mimic the action of GLP, an incretin hormone that increases insulin secretion whilst inhibiting glucagon release. 71 They are approved for use in the treatment of patients with type 2 diabetes mellitus and have been shown in pre-clinical studies to promote β-cell proliferation and reduce β-cell apoptosis. 72 In WS patients, treatment with GLP-1R agonists reduces insulin requirements and optimizes glycemic control.73,74 In cell models of WS, GLP-1R agonists provide a mechanism for β-cell adaptation to metabolic and cellular stress through the PERK-ATF4 pathway. 75 In a rat model of WS, preventative treatment before the onset of metabolic symptoms with the GLP-1R agonist liraglutide reduced ER stress and inflammation. 76 The neuroprotective benefit of liraglutide in patients with WS is currently under evaluation. 77
Inhibitors of dipeptidyl peptidase-4 (DPP-4), an enzyme that deactivates GLP-1 and increase GLP-1 levels, have been studied in cell models. Sitagliptin, a DPP-4 inhibitor approved for use in adults with type 2 diabetes mellitus, was found to significantly decrease β-cell death in rat INS-1-E cells treated with thapsigargin, by stabilizing cellular calcium levels and decreasing levels of the pro-apoptotic protein thioredoxin-interacting protein. 78
AMX0035 and mesencephalic astrocyte-derived neurotrophic factor (MANF) are two other molecules that are being investigated as potential therapeutic targets for WS by modulating ER stress. Previous studies have shown that loss of MANF in vivo leads to β-cell death with ER stress. 79 In cell and mouse models of WS, MANF-based treatments have been shown to enhance β-cell proliferation and prevent β-cell death. 80 The mechanism is not fully understood and the receptors for MANF have not been identified yet, but it is likely due to suppression of the proapoptotic arm of ER stress signaling. A neuroprotective gene therapy strategy involving direct delivery of MANF to affected cells using adeno-associated virus (AAV) delivery systems is being contemplated. 81
Modulating mitochondrial function
Compounds that optimize mitochondrial function are being investigated in a range of conditions that lead to primary or secondary mitochondrial dysfunction, including glaucoma and late-onset neurodegenerative disorders such as Alzheimer disease.82 –84 Nicotinamide adenine dinucleotide (NAD+) is an essential redox molecule that acts as a co-substrate for several enzymes regulating crucial metabolic processes, including the tricarboxylic acid cycle, fatty acid oxidation, and mitochondrial oxidative phosphorylation. NAD+ is also involved in DNA repair pathways, calcium homeostasis, and microtubular organization in neuronal cells.85 –90 Two critical enzymes that consume NAD+ as a substrate are sirtuins and poly (ADP-ribose) polymerases (PARPs). Sirtuins are NAD+-dependent deacetylases that play a key role in transcription regulation, DNA repair, and resistance against oxidative stress. 91 There are seven mammalian sirtuins with diverse localization, enzymatic activity, and protein substrates. NAD+ acts as a co-substrate for protein deacetylation, which is an important regulatory mechanism for optimal mitochondrial function, including processes such as fatty acid oxidation and the production of ketone bodies. 90 NAD+ is also an important substrate for PARPs, which are important for DNA repair, epigenetic modification, cell differentiation, and adaptive responses to inflammatory and oxidative stress. 91 PARPs hydrolyze NAD+ to transfer an ADP-ribose moiety to a receptor amino acid. Although PARP activation is an integral part of the cellular response to oxidative stress, PARP overactivation can lead to overconsumption of cytosolic NAD+, glycolytic inhibition, and cell death. 91
Activation of the NAD+-dependent pathway has been shown to improve mitochondrial function by mitigating the effects of reactive oxygen species (ROS),92,93 thereby protecting cells from oxidative damage and induced apoptosis. 89 Disrupted NAD+ homeostasis is involved in stem cell senescence and impaired capacity for tissue maintenance and regeneration. 94 Restoring NAD+ levels could, therefore, enhance the cell’s defense against oxidative stress and neurodegeneration. Increasing NAD+ availability by supplementation with NAD+ precursors or intermediates, or by inhibiting NAD+-consuming enzymes such as PARP, have been shown to increase sirtuin activity conferring neuroprotective benefits. 89
Animal models of mitochondrial disease show improved mitochondrial function by increasing NAD+ availability. 95 In a mouse model of glaucoma, increasing retinal NAD+ levels with dietary nicotinamide (NAM), a precursor of NAD+, or expression of the gene encoding for a NAD-producing enzyme (Nmnat1) provided robust neuroprotection of RGC, with preservation of RGC function and reversal of age-related transcriptomic changes.96 –98 The neuroprotective effects of nicotinamide were also observed in rodent models of axon degeneration and mitochondrial degenerative insults. 99 In a crossover, double-masked, clinical trial of 57 glaucoma patients randomised to oral placebo or NAM for 12 weeks, NAM supplementation led to an early improvement in inner retinal function as determined by electroretinography. 83 This is consistent with NAD+ potentially delaying the progression of glaucoma by compensating for the age-related decline of NAD+ levels and mitochondrial biogenesis in RGCs. 96 Although the exact molecular mechanisms were not defined, niacin (vitamin B3), which is a precursor for the synthesis of NAD+, also improved muscle performance in patients with mitochondrial myopathy. 93 As a result, based on the growing body of evidence that NAD+ precursors can block neurodegeneration under conditions of compromised mitochondrial function, supplementation with niacin or NAM could improve RGC health and preserve vision in WS patients.
Idebenone has been intensively investigated for its antioxidant properties in patients with mitochondrial disease.100,101 It is a short-chain benzoquinone (2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinone) that is hydro soluble compared with coenzyme Q10 (CoQ10), giving it the ability to penetrate the blood-brain barrier. Idebenone is the only medication that has been approved for the treatment of LHON.99,102 By increasing the efficiency of electron flux along the mitochondrial respiratory chain, idebenone maximizes ATP production and decreases the production of ROS with an inhibitory effect on apoptosis. 103 In addition to being an efficient electron carrier that allows a compromised complex I to be bypassed in LHON, idebenone also acts as an inhibitor of lipid peroxidation.104,105 In one case report, a patient with WS carrying a confirmed WFS1 mutation experienced progressive subjective visual improvement following treatment with idebenone over a period of 6 months. 106 A retrospective case series of patients with another mitochondrial optic neuropathy, autosomal DOA caused by pathogenic OPA1 mutations, found that idebenone could stabilize visual acuity. 107 Although encouraging, these findings should be viewed with caution, and adequately powered treatment trials using standardized treatment doses and follow-up protocols are required to evaluate the benefit of idebenone in WS and DOA caused by WFS1 and OPA1 mutations, respectively.
Gene therapy
Two strategies for gene therapy that represent promising therapeutic possibilities in WS include the use of AAV systems to transfer the wild-type WFS1 gene into RGCs and the use of CRISPR gene-editing technology to correct the mutation in iPSCs. The eye is an ideal target organ for gene therapy, as the various retinal layers can be accessed directly and it benefits from relative immune privilege. 108 Voretigene neparvovec was the first gene therapy product approved for the treatment of a genetic eye disease, namely Leber congenital amaurosis caused by biallelic RPE65 mutations. 109 Several trials are investigating the use of gene therapy in other inherited forms of outer retinal degeneration and optic neuropathy, in particular LHON, which is the focus of several phase III clinical trials.110 –112 The published results indicate significant visual improvement in LHON patients with the m.11778G>A mtDNA mutation, treated within 1 year of disease onset with an intravitreal injection of recombinant AAV2/2-ND4.110,111 An attractive strategy for WS would be to replace the defective gene in RGCs, especially in patients carrying recessive WFS1 mutations. Although additional preclinical work is required before the initiation of human trials an AAV2-CMV-WFS1, vector injected intravitreally has shown stabilization of visual acuity at three- and six-months post-injection in a mutant mouse model (Wfs1exon8−/−). 113
The ability to transform post-mitotic cells, such as fibroblasts, into iPSCs and the development of gene editing technologies, have opened up new therapeutic opportunities for genetic diseases. CRISPR-Cas9 technology has been used to correct the WFS1 pathogenic variant in iPSCs that were reprogrammed from a patient’s fibroblasts. These iPSCs were then used to generate autologous, gene-corrected pancreatic β-cells, which remarkably exhibited dynamic glucose-stimulated insulin release and expressed similar cell markers when compared with the patient’s native unedited pancreatic β-cells. 114 Mice with pre-existing diabetes that were transplanted with gene-corrected β-cells demonstrated improved glucose tolerance and they maintained blood glucose normalization over the six-month observation period. Gene-correction also reversed the effects of cellular stress observed in unedited cells that had been treated with thapsigargin. 114 These findings are exciting, but there are number of challenges in relation to ocular gene therapy for WS. Unlike the relatively simple environment of pancreatic β-cells, RGCs not only need to be transplanted in the correct anatomical position, but their axons will need to migrate along the optic nerve and establish accurate retinotopic connections with the lateral geniculate nucleus. Due to the limited availability of retinal and optic nerve tissues from patients with WS, iPSC-derived RGCs could be used to study the effects of WFS1 mutations in a more complex neuronal system and importantly, provide a valuable resource for drug screening.
Barriers to translational research
Considerable progress is being made in understanding the mechanisms that underpin the pathophysiology of WS, leading to the development of several therapeutic strategies outlined above. In addition, greater awareness of WS and increasing recognition of the spectrum of phenotypes that exist, have been used to generate research foci for basic science, using a ‘bench to bedside and back to bench’ approach. However, several barriers to translational research exist at all levels of the therapeutic pipeline, slowing progress in the delivery of benefits to individuals with WS.
Research into WS has been limited by its rarity and the inability to get access to eye and brain tissues from affected patients. Furthermore, WS is clinically heterogeneous and the correlation between genotype and phenotype remains poorly understood. Although several animal models exist for WS-1 and WS-2, efficient in vivo model systems that faithfully recapitulate the clinical and pathological hallmarks of human WS are lacking, particularly for the study of the neurological and ophthalmological manifestations of WS. In addition, the study of RGCs and other neuronal populations in their native environment is challenging due to their complex interactions with other cell types. The use of iPSCs to generate 3D retinal organoids provide a tractable solution, but the yield of RGCs is variable due to their tendency to be lost during organoid maturation. 115
Like many rare diseases, clinical research in WS face barriers relating to funding and the need for multinational collaborations. The establishment of patient-organized networks such as Wolfram Syndrome UK in the United Kingdom, as well as international organizations such as The Snow Foundation and Wolfram Syndrome Research Alliance (WSRA), have helped to develop an infrastructure for patients, scientists, and clinicians to address the challenges faced by individuals with WS. In addition, registries such as the Wolfram Syndrome International Registry (Washington University in St. Louis), the Wolfram Syndrome Global Registry (The Snow Foundation), and the European Wolfram Syndrome Registry (Euro-WABB project) provide a tool for tracking the clinical care and natural history of individuals with WWS and provide a platform for recruiting patients into clinical trials. However, the clinical heterogeneity of WS and the lack of a universal system for classifying and diagnosing WS can make recruitment of patients into clinical trials challenging.
The research and development journey of new drugs that make it to market is long and costly. Drug repurposing represents an opportunity for rare diseases; it is often presented as being a viable, risk-managed strategy for pharmaceutical companies developing orphan drugs.116,117 Although there are notable successes, drug repurposing does not always succeed. A major reason is the failure of the repurposed drug to demonstrate a benefit-risk profile in clinical trials that would support regulatory approval for the new indication. The overall cost of drug development may also be increased by the need to conduct trials to establish the dose-response relationship in the new indication. Crucially, the financial commitments of drug repurposing may not be commercially attractive for companies, particularly for drugs for which generic versions are already available.
Conclusion
WS is a progressive neurodegenerative disease that affects multiple organs, resulting in significant morbidity, in particular irreversible neurological deficits and blindness. The clinical manifestations of WS reflect the failure of specific cell populations, for example, insulin-dependent diabetes mellitus and optic atrophy arising from the loss of pancreatic β-cells and RGCs, respectively. The sustained research effort that has been invested in the field since the identification of WFS1 as the major gene causing WS in 1998 has helped build a clearer picture of how lack of wolframin results in neurodegeneration and the development of disease. The use of both in vivo and in vitro model systems of WS have highlighted the importance of calcium dysregulation, ER stress, and mitochondrial dysfunction in precipitating apoptosis. However, further work is needed to define the precise molecular targets and how WFS1 mutations can result in such variable phenotypes and disease severity. Although there are currently no proven effective treatments for WS, a number of complementary therapeutic strategies are being explored, including drug repurposing and gene therapy. We have now entered an exciting and fast-moving translational phase for WS that will hopefully lead to the long-awaited breakthroughs for patients and their families. The road ahead will be challenging: this effort will require close and concerted collaboration between research groups, industrial partners, and patient-led organizations.
Footnotes
Author Contributions
Ratnakar Mishra: Formal analysis; Methodology; Writing-original draft; Writing-review & editing
Benson S Chen: Formal analysis; Methodology; Writing-original draft; Writing-review & editing
Prachi Richa: Formal analysis; Writing-review & editing
Patrick Yu-Wai-Man: Conceptualization; Formal analysis; Methodology; Writing-original draft; Writing-review & editing
Conflict of interest statement
The authors declare that there is no conflict of interest.
Ethics statement
Ethical approval and informed consent were not required for this review.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: BSC is recipient of the Cambridge-Rutherford Memorial Scholarship awarded by the Royal Society Te Apārangi – Rutherford Foundation and the Cambridge Commonwealth, European & International Trust, and also the Aotearoa New Zealand Fellows Research Entry Scholarship awarded by the Royal Australasian College of Physicians (RACP). PYWM is supported by a Clinician Scientist Fellowship Award (G1002570) from the Medical Research Council (UK), and also receives funding from Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity, the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the International Foundation for Optic Nerve Disease (IFOND), the UK National Institute of Health Research (NIHR) as part of the Rare Diseases Translational Research Collaboration, the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014), and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.