
Abstract
Background
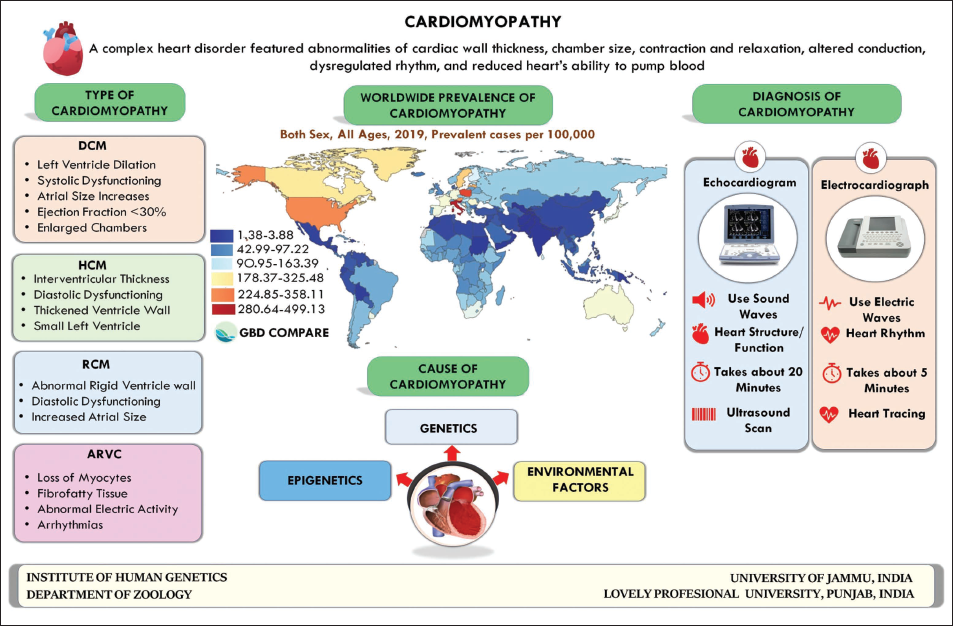
Cardiomyopathy, a rare heart disease, is characterized by abnormalities in cardiac wall thickness and chamber size, leading to impaired contraction, relaxation, conduction, rhythm, and reduced pumping ability.
Aim
This review aims to provide a comprehensive understanding of cardiomyopathy by examining its various aspects
Method
A literature survey was conducted using online databases such as PubMed, Google Scholar, and Web of Science, covering publications from January 1995 to July 2023.
Result
Genetic mutations in key muscle contraction genes (MYH7, MYL2, MYL3, MYBPC3, TNNT2, TPM1, TNNI3, ACTC) contribute to cardiomyopathy. Additionally, epigenetic markers in genes like FKBP5, TBX5, HAND1, POLA2, PLAAT3, and CCDC88B, along with environmental factors such as alcohol addiction, smoking, and stress, significantly influence disease risk. Genetic testing, including whole exome/genome sequencing, has revolutionized diagnosis, enabling early detection and intervention. Familial genetic testing facilitates personalized management.
Conclusion
Cardiomyopathy is a complex disease with genetic and environmental influences. Various techniques, including genetic testing, aid in its identification and management. Furthermore, machine learning (ML) techniques have emerged as valuable tools in understanding and predicting cardiomyopathy outcomes.
Introduction
It is well recognized that the heart is the first organ to form in an embryo, and the development of the heart has a significant impact on the development of succeeding organs. However, heart disease poses a significant threat as a leading global cause of morbidity and mortality. This broad category includes conditions like congenital heart defects, valvular diseases, arrhythmias, heart failure, coronary artery disease, and cardiomyopathy. Cardiomyopathies, in particular, are complex disorders marked by altered heart chamber size, abnormal cardiac wall thickness, disturbed conduction, impaired relaxation and contraction, dysregulated rhythm, and reduced pumping ability. 1 The term “cardiomyopathy” was first introduced in 1957 by Wallace Brigden at the National Heart Hospital in London to describe a group of basic myocardial diseases. Subsequently, the WHO used the term to refer to heart diseases with no known cause. In 2006, the American Heart Association developed a modern classification system based on recent advancements, categorizing cardiomyopathies into hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC), and restrictive cardiomyopathy (RC). 2
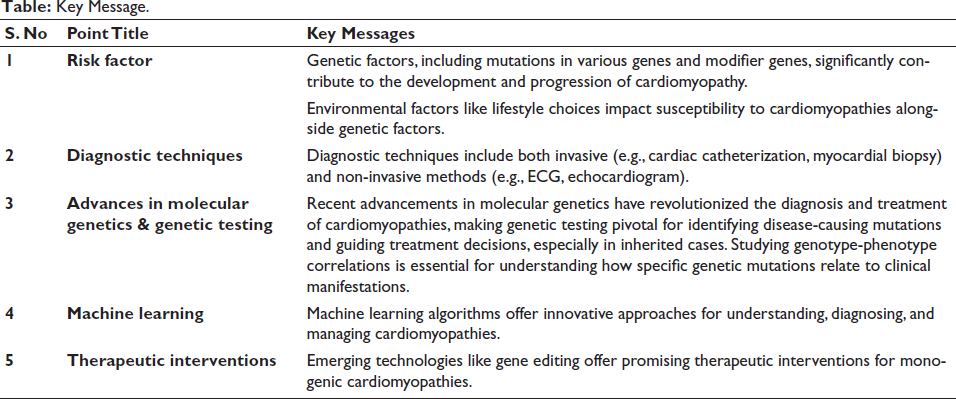
The risk of heart disease is influenced by various factors, broadly grouped into genetic and environmental categories. Genetic factors, involving numerous genes (polygenic), affect disease susceptibility. Environmental factors further increase the likelihood of developing these diseases. Over the past two decades, research into the causes of cardiomyopathies has progressed significantly, leveraging human and model organism molecular genetics and proteomics.3, 4 Given these advancements, this review aims to explore several aspects of cardiomyopathy, including its clinical types, the risk factors that increase susceptibility, diagnosis, and therapy. The ultimate objective is to provide a comprehensive understanding of the complexity of cardiomyopathy.
Method
By utilizing keywords such as cardiomyopathy, polymorphism, genetics, environment factors HCM, DCM, genetic variants, association study of cardiomyopathy, environmental factors and cardiomyopathy, diagnostic of cardiomyopathy, treatment of cardiomyopathy, etc. we searched Google Scholar, PubMed, Springer, and Elsevier’s online databases for research publications published between January 1995 and July 2023. The literature search and analysis were done by our four authors (S.B., A.S., K.M., S.S.), and any conflict between the authors was resolved by another author (P.K.).
Background
Cardiomyopathy is a rare, complex, and heterogeneous disease and is linked with electromechanical dysfunction, which typically exhibits improper ventricular dilation or hypertrophy. Concerning the statistics of the disease, Global Burden Diseases-2019 (
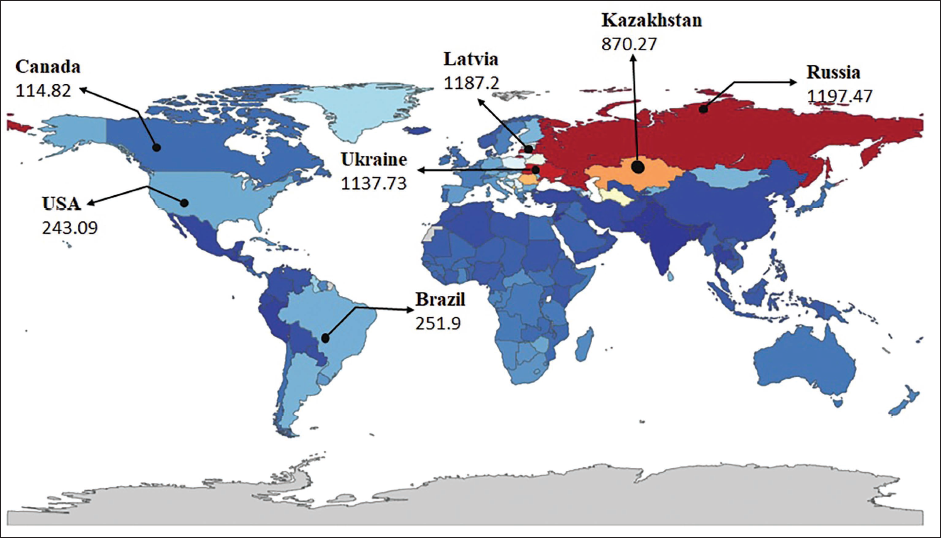
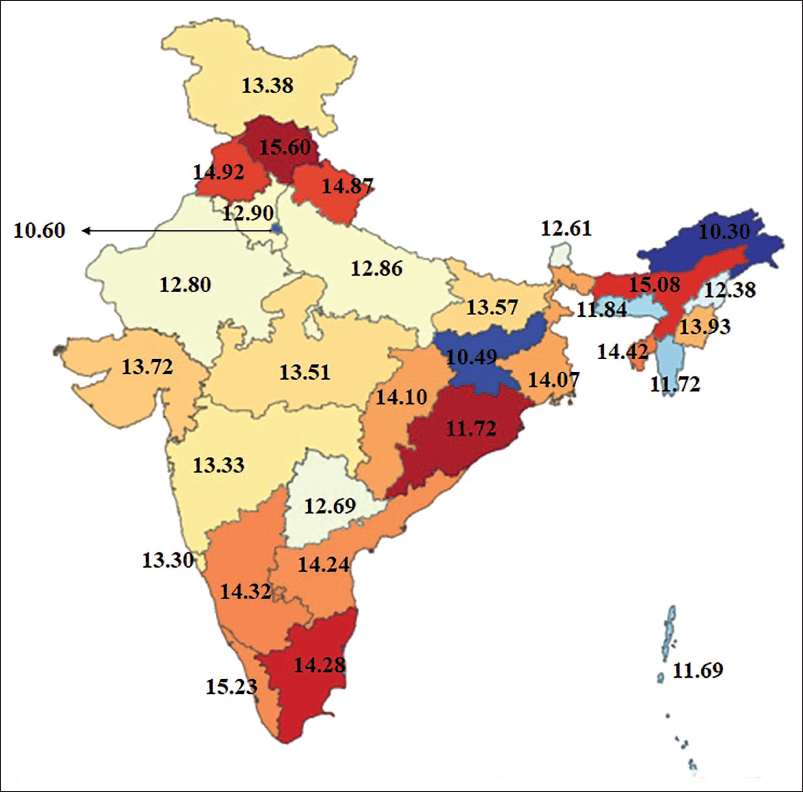
Types of Cardiomyopathies
Recent advances in genomics and proteomics have increased our understanding related to the disease, resulting in the categorization into four major types such as DCM, HCM, RC, and ARVC. 6 Such classification makes this area of study clearer and makes it easier for medical and scientific groups to work together on different aspects ranging from identification of the risk variable to disease diagnosis, predicting, and treating this complicated disorder (CVDs) (WHO.INT).
Hypertrophic Cardiomyopathy
HCM has been extensively studied since its initial description in 1869. 7 Advances in clinical approaches have enhanced our understanding of this condition. HCM is an autosomal dominant disorder affecting about 1 in 500 adults and is a leading cause of sudden cardiac death in those under 50, especially young athletes. It is characterized by excessive cardiac muscle thickness, which impairs the heart’s ability to pump blood efficiently. 8
Dilated Cardiomyopathy
Heart abnormalities that begin in the left ventricle (LV) and progress to impact the right ventricle characterize DCM, the second most common form of cardiomyopathy. Enlargement and weakness of the ventricles cause the heart to lose its ability to pump blood effectively, which can manifest as a variety of conditions including irregular heart rhythm, heart valve disease, heart failure, and blood clots in the heart. 9
Restrictive Cardiomyopathy
RCM is a rare form of cardiomyopathy characterized by stiff ventricular walls, leading to abnormal ventricular activity, including an inability to relax and fill properly. As the condition worsens, valve problems intensify, heart muscle weakness accelerates, and the ventricles lose their pumping function, resulting in heart failure. Symptoms of left or right ventricular failure may arise, affecting one or both ventricles. RCM should be suspected when heart failure presents without cardiomegaly or systolic dysfunction. Clinically, cardiac sarcoidosis is linked to RCM in 5% of cases, presenting with tachyarrhythmias, heart block, syncope, and sudden death. Additionally, cardiac amyloidosis replaces normal heart muscle with amyloid deposits, disrupting function and electrical signals, leading to RCM. This progressive condition, caused by genetic variants or acquired conditions, is often discovered incidentally. 10
Arrhythmogenic Right Ventricular Cardiomyopathy
ARVC, another rare form of cardiomyopathy that used to be called arrhythmogenic right ventricular dysplasia, is another type of cardiomyopathy that is marked by degeneration of the right ventricle and irregular heartbeats in the ventricle. 11 ARVC is characterized by the fatty or fibrous tissue replacing the right ventricle’s muscular tissue, leading to disruptions in the heart’s electrical signals and, in extreme cases, sudden cardiac arrest. 12
Various types of cardiomyopathy, including left ventricular noncompaction cardiomyopathy (LVNC), characterized by trabeculations, enlargement, and altered dilation in the LV, 13 can lead to malignant ventricular arrhythmias, exacerbated by intense physical exercise, due to myocyte mechanical disconnection increasing pressure, afterload, and wall stress in the RV more than the LV. 14
Alcoholic Cardiomyopathy
Alcoholic cardiomyopathy stems from chronic, heavy alcohol use, with even lower amounts posing risks to susceptible individuals. Direct toxic effects of alcohol and its metabolites, like acetaldehyde, damage heart muscle cells, leading to structural and functional heart alterations. 15 While historical debates existed, research increasingly emphasizes alcohol’s direct toxic effects on cardiac and skeletal muscle. 16 Studies confirm alcohol’s dose-dependent toxicity to both cardiac and striated muscle, highlighting its role in cardiomyopathy development. 17
Both acute and chronic alcohol use decrease myocardial contractility through mechanisms involving the activation of the neurohormonal system. 18 The multifaceted causal mechanism appears linked to the production of reactive oxygen species, mitochondrial damage, and decoupling of the electron-transport chain.18, 19 Histologic findings of alcoholic cardiomyopathy closely resemble idiopathic DCM, with high cardiac output failure as a distinguishing feature. Diagnosis requires ruling out other causes alongside heavy alcohol intake history. Even lower alcohol amounts can lead to cardiomyopathy in susceptible individuals. Understanding the direct mechanisms of alcohol-induced cardiomyopathy is crucial for management and prevention. Healthcare providers, armed with this knowledge, can emphasize alcohol’s toxic effects on heart muscle cells, urging alcohol cessation and lifestyle modifications. Dexrazoxane shows promise as a cardioprotective measure by reducing free radical production. Exploring non-invasive and serological methods for early detection could improve prognosis. 18 Therefore, preventing alcoholic cardiomyopathy involves limiting or abstaining from alcohol, coupled with education about its risks and help-seeking for alcohol use disorders. 15
Reversible Cardiomyopathy
Reversible cardiomyopathy refers to a condition where the heart muscle experiences dysfunction that can be improved or reversed with appropriate treatment. Unlike irreversible cardiomyopathies, which lead to permanent damage or progressive deterioration of heart function, reversible cardiomyopathies offer the potential for recovery and restoration of normal cardiac function. 20 Various factors can contribute to reversible cardiomyopathy, including metabolic imbalances, nutritional deficiencies, inflammatory processes, or exposure to toxins. Identifying and addressing the underlying cause of cardiac dysfunction can lead to the restoration of normal heart function in reversible cardiomyopathies. Some reversible cardiomyopathies that can potentially return to normal cardiac function with appropriate management include:
Tachycardia-induced Cardiomyopathy
Correction of the underlying arrhythmia leading to rapid heart rate can improve cardiac function and reverse the effects of tachycardia-induced cardiomyopathy. 21
Peripartum Cardiomyopathy (PPCM)
It is a rare heart condition occurring during or shortly after childbirth. 22 It typically presents with heart failure symptoms and LV systolic dysfunction in the third trimester or within five months post-delivery. Risk factors include age, obesity, and a history of preeclampsia, especially in women over 30 with multiple pregnancies. PPCM weakens the heart muscle, impairing its pumping function and causing heart failure symptoms. Diagnosis can be challenging due to overlapping symptoms with normal pregnancy discomforts, but timely recognition is crucial for effective management. 23 Treatment involves medications like angiotensin-converting enzyme inhibitors and diuretics, along with close monitoring. With proper care, including medication and monitoring, PPCM can improve, and cardiac function may return to normal in some cases. 20
Inflammatory Cardiomyopathy
Addressing the underlying inflammatory processes through medications and therapies can help reverse the effects of inflammatory cardiomyopathy and restore normal heart function. 20
Hyperthyroidism-induced Cardiomyopathy
Treating the underlying hyperthyroidism with medications, radioactive iodine therapy, or surgery can lead to the reversal of hyperthyroidism-induced cardiomyopathy. 24
Takotsubo Cardiomyopathy
Also known as stress-induced cardiomyopathy (SIC) or broken heart syndrome, is a reversible heart condition characterized by sudden and temporary weakening of the heart muscle. It is typically triggered by intense emotional or physical stress, such as the death of a loved one or a traumatic event. While it predominantly affects postmenopausal women, it can occur in individuals of any age or gender. 25 The exact mechanisms behind Takotsubo cardiomyopathy are not fully understood but likely involve complex interactions among neurohormonal, vascular, and myocardial factors. Treatment usually involves supportive care, medications, and lifestyle changes, often leading to the restoration of normal cardiac function. 26
Chronic Illness-induced Cardiomyopathy
Managing underlying chronic diseases such as cirrhosis, obesity, or kidney failure can help improve cardiac function and reverse the effects of chronic illness-induced cardiomyopathy. 27 These reversible cardiomyopathies highlight the importance of identifying and addressing the underlying causes to facilitate the restoration of normal cardiac function through appropriate management strategies.
Risk Factors
Risk factors are defined as the “factors responsible for altering the susceptibility of diseases/lowering the threshold of disease’s susceptibility.” 28 Over the previous passing years, several risk factors have been discovered, although describing them together might be difficult therefore, they are broadly categorized into genetic and environmental risk factors.
Genetic Factors
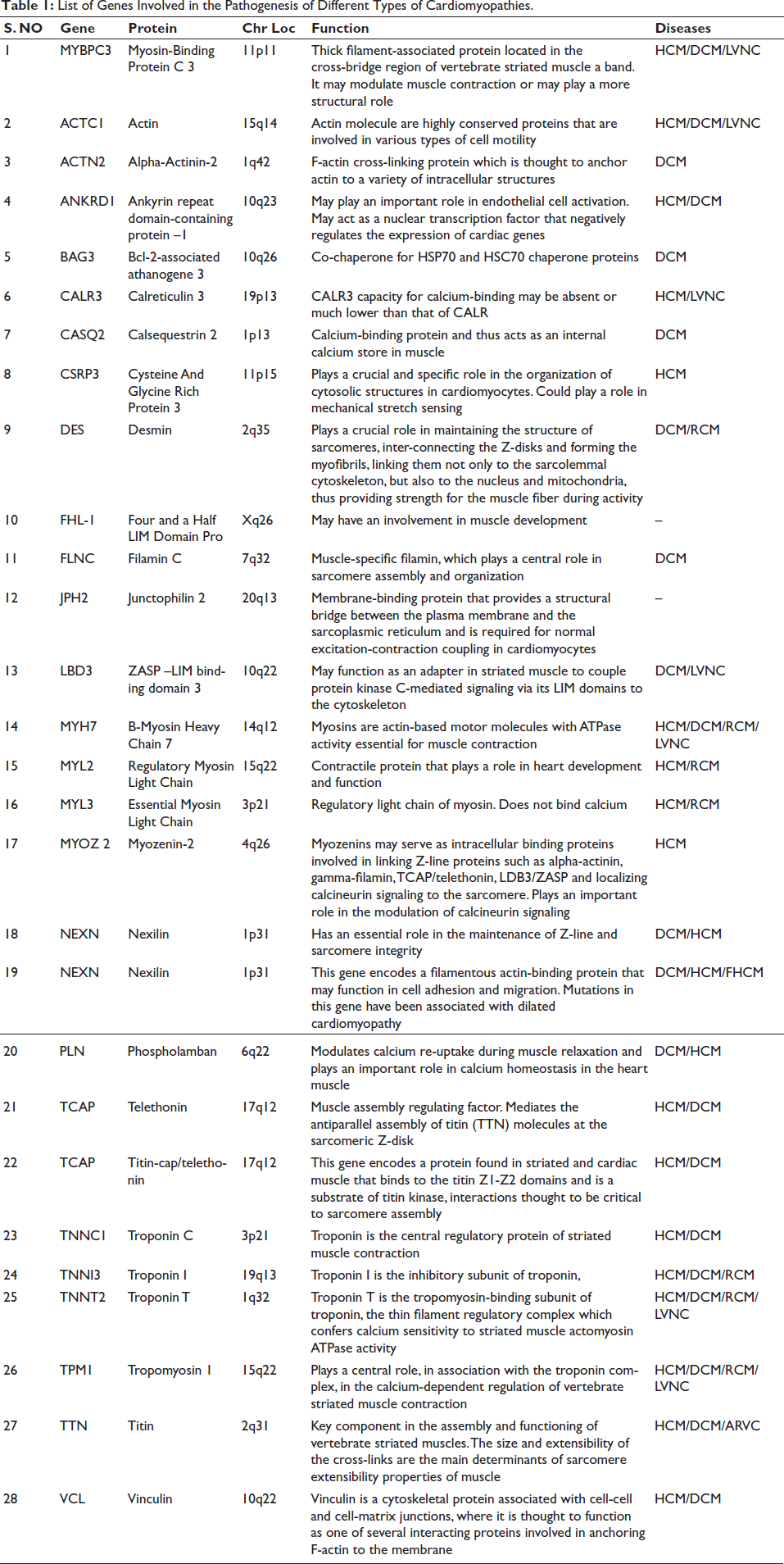
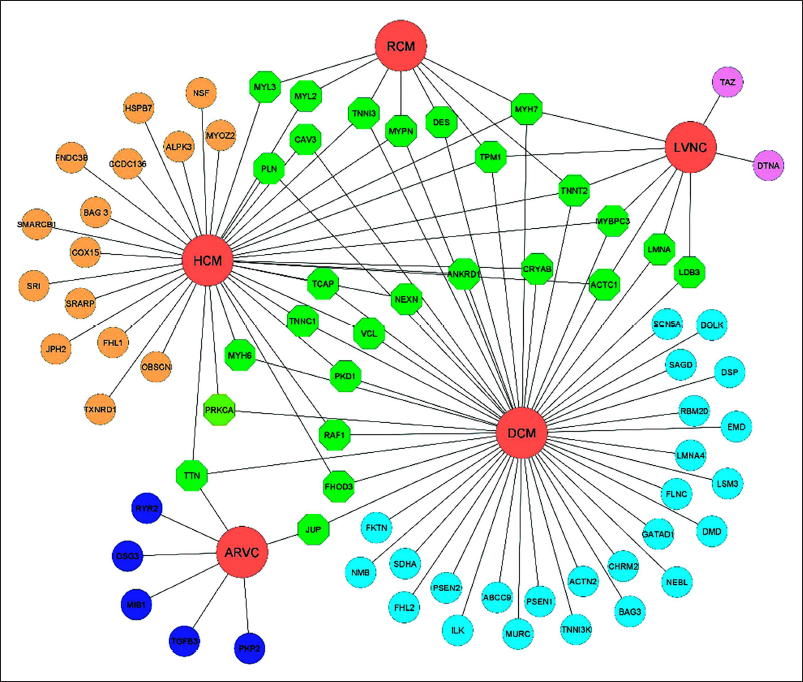
In most cases, a genetic condition can be traced back to a change in one or more genes, and numerous genes have been discovered to exhibit de novo mutations, which are novel mutations not present in either biological parent (Table 1). Cardiomyopathy is not directly caused by mutations in genes that regulate heart function, but many other genes, called modifier genes, may play a significant role in the development and progression of the condition. Mutations such as deletion or insertion may result in a protein of altered size, and insertion of an early termination codon may also lead to frameshift mutations. 29 Regardless of the type of mutation, they are responsible for almost three-quarters of cardiomyopathies.
List of Genes Involved in the Pathogenesis of Different Types of Cardiomyopathies.
Key Message.
Recent advancements in molecular genetics have significantly enhanced our understanding of disease pathophysiology at the molecular level. Crucial genes involved in the modification and regulation of muscle contraction, such as beta-myosin heavy chain (MYH7), MYL2 (regulatory myosin light chain), MYBPC3 (myosin-binding protein C), MYL3 (essential myosin light chain), TNNI3 (cardiac troponin I), TPM1 (alpha-tropomyosin), TNNT2 (cardiac troponin T), and ACTC (actin), have been identified. Mutations in these essential sarcomeric genes commonly underlie the most frequent genetically induced form of HCM, known as Myofilament-HCM. Specific mutations in these genes, such as beta-MHC (Arg869Gly) and cardiac troponin T (Ser179Phe), are associated with an extremely severe phenotype known as homozygous HCM. This phenotype typically manifests in childhood and is characterized by myocardial fibrosis, severe hypertrophy, and a higher frequency of sudden cardiac death (SCD), contributing to early mortality.30, 31 Clinical features of HCM are often defined by mutations in MYH7, impacting cardiac force generation, LV hypercontractility, and stiffness. MYBP-C mutations can slow myofibril contraction by influencing sarcomere organization. 32 Notably, genes such as TNNI3, TNNT2, TPM1, and MYL3 are less frequently involved than β-MYHC and MBP-C in HCM patients. 33 Genotype-phenotype studies reveal that different HCM disease genes have distinct time frames for symptom development. For instance, individuals with a mutation in the β-MHC gene typically exhibit symptoms in their early twenties, while those with a mutation in the cMyBP-C gene may not show symptoms until their late fifties or early sixties. 34 However, genotype-positive individuals may remain asymptomatic or experience symptoms such as lightheadedness, palpitations, syncope, and sudden death.
Concerning another common form of cardiomyopathy, DCM, numerous genes are implicated in its pathogenesis, leading to a high level of locus heterogeneity. These genes include those encoding nuclear lamina proteins (LMNA), dystrophin-associated glycoprotein complex, sarcomeric proteins, intermediate filaments, and Z-disks.35, 36 Additional genes associated with sporadic and familial DCM have been identified, such as BAG3 (BCL2e Associated Athano Gene-3), RBM20, and TTN, with TTN responsible for up to 25% and 18% of familial and sporadic DCM, respectively.37, 38 Titin, positioned at both ends of the sarcomere from the Z-line to the M-line, regulates sarcomere length and assembly. Mutations in the TTN gene lead to aberrant titin synthesis, affecting sarcomere shape and function during diastolic filling by passively elongating the muscle and regulating active contractile force. 39 The chaperone protein encoded by the RBM20 gene, known as “Ribonucleic Acid Binding Motif-20,” binds to mRNA and controls exon splicing to produce various gene isoforms, including titin. RBM20 mutations, along with TTN mutations, can result in DCM.40, 41 Additionally, FLNC, a cytoskeletal actin-binding intermediate filament, serves as a linker between membrane proteins and sarcomeres, contributing significantly to sarcomere stability. Z-disc proteins, for example, crosslink actin filaments to form a three-dimensional actin web, binding them to membrane proteins and ion channels, thereby enhancing sarcomere and Z-disk structure stability.42, 43
RCM is generally heterogeneous, exhibiting multiple inheritance patterns such as autosomal dominant, recessive, X-linked, or mitochondrial transmission. The majority of the genes discovered encode sarcomeres or Z-disk proteins, such as TTN, MYL3, MYH7, TNNT2, ACTC1, TPM1, TNNI3, MYL2, DES, MYPN, BAG6, 44, 45 and TTR. It is essential to take note of the fact that the TTR gene and the DES gene have been discovered to be associated with amyloid-related RCM and desmin-related myopathy, skeletal myopathy, and/or atrioventricular block, respectively. 44
Researchers have also looked for mutations in AVRC 46 and found multiple genes such as PKP2 (Plakophilin-2), DSP (Desmoplakin), DSG3 (desmoglein) which are components of the desmosomal complex and are important for electricity conductance. 47 When it comes to mechanical stability and electrical coupling between myocardial cells, desmosomal structures are crucial since they are responsible for making the connection. Thus any change in the desmosomal complex would ultimately alter myocardial activity.48, 49 In addition, nuclear signaling and transcriptional activity, especially those mediated by beta-catenin-controlled pathways, have also been demonstrated to be affected by desmosomal change, for example, increased expression of adipogenic and fibrogenic genes leads to the development of fibro-fatty myocardial scarring. 43 According to Tiso and group, RYR2 (ryanodine receptor 2) which is an important regulator of sarcoplasmic calcium ions has been found with many mutations in ARVC patients, which connected cardiomyopathy to channelopathy. 50
After analyzing multiple genes for different types of cardiomyopathy a complex gene-gene interaction between the candidate genes of cardiomyopathy was formed using the String database (STRING: functional protein association networks (

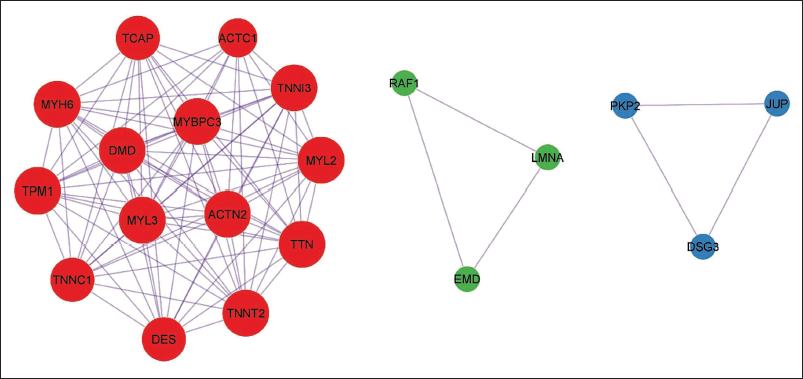
In the concluding part of the present section, it was observed that the susceptibility to cardiomyopathy is determined by the interplay of several genes that are shared among different forms of cardiomyopathy.
Epigenetic Marks
The notion that diseases can be caused by inherited marks rather than changes in gene sequences has revolutionized disease understanding, ushering in the era of epigenetics. Epigenetic studies focus on regulatory mechanisms and functional repercussions of altered gene expression, including non-coding RNAs, chromatin remodeling, DNA methylation, and histone modifications. Any alteration in these mechanisms can increase disease occurrence, making epigenetic regulation pivotal in human physiology and pathology. Scientists increasingly view epigenetics as a more intriguing risk factor for cardiomyopathies than gene mutations. Factors such as methylation and acetylation can directly affect gene expression, while early-life experiences and substance exposure can influence gene expression through epigenetic mechanisms. Many epigenetic factors are explored which include high mRNA expression of genes like LY75 (Lymphocyte antigen 75) and ADORA2A (Adenosine receptor A2A) are associated with altered DNA methylation in patients suffering from DCM. 51 Also, patients with DCM have aberrant epigenetic patterns that cause several metabolites involved in glycolysis and the citric acid cycle to be increased in the serum by as much as 5.7-fold. 52 Casares-Marfil and group found different genes such as POLA2, PLAAT3, and CCDC88Bas a significant cis methylation quantitative trait locus within chromosome 11 associated with Chagas cardiomyopathy. 53 Specific CpG hypomethylation in the FKBP5 gene responsible for inflammatory response is found to be correlated with the pathogenesis of DCM. 54 Watanabe and group observed that patients suffering from DCM have 5-methylated cytosine cardiomyocytes which significantly inversely correlated with LV functional parameters such as heart rate, end-diastolic pressure, and ejection fraction and a positive correlation with LV dilatation and volume index at systole and LV remodeling parameters (LVH). 55 Altered CpG methylation in the LAD gene (Lamin Associated Domain) which encompasses 20% of the human cardiac myocyte genome is associated with altered expression of gene. 56 Jo and colleagues observed alterations in DNA methylation in the cis-regulatory regions of cardiac development genes such as TBX5 and HAND1, which are involved in ventricular development. 57
Environmental Factors
Despite the huge impact of genetic factors on susceptibility to diseases, environmental factors also known as non-genetic factors include various categories of factor classes such as lifestyle, socio-demography factors, etc. Environmental epidemiological studies have been continuously searching for decades about the different environmental risk attributes and found many factors such as stress, diet, smoking & alcohol consumption, etc.
Smoking
Smoking, an omnirisk factor, not only increases the risk of cardiac disorders but also various other conditions, affecting LV wall-motion abnormalities. Long-term cigarette smoking can lead to “smoke cardiomyopathy,” characterized by significant metabolic and structural changes in the heart muscle. 58 Smokers have a higher risk of non-cardiac death in older age groups and cardiac death in middle-aged adults. Smoking alters heart function, leading to tachycardia at rest and during exercise, reducing vagal modulation, and increasing sympathetic dominance. 59 Hendriks and group 60 demonstrated a significant correlation between smoking and increased left ventricular (LV) and right ventricular (RV) end-systolic volume, reduced LV peak measures, and impaired global longitudinal, radial, and circumferential strain, along with reduced LV and RV ejection fraction. Tobacco smoke exposure is also associated with myocardial hypertrophy, LV enlargement, and ventricular dysfunction, mediated by changes in hemodynamics, neurohormones, oxidative stress, inflammation, and matrix metalloproteinases. 61 Continued smoking after a heart transplant significantly reduces survival rates due to the accelerated development of graft vasculopathy and cancer. Smoking cessation should be emphasized early to prevent such heart alterations and early cardiac death. 62 Factors associated with quitting smoking include younger age, higher weight, lower anxiety and depression levels, employment status, partnership, and lower risk status. 63
Alcohol
Alcohol consumption is linked to various conditions, including cardiomyopathy, where chronic heavy drinking significantly increases the risk of disease. Moderate to excess alcohol intake, compared to moderate consumption, adversely affects heart functionality and muscle strength, leading to increased chamber dilation (left atrium and ventricle, and right ventricle), reduced biventricular function, and increased LVH. 64 Stopping alcohol consumption can lead to significant improvement in ejection fraction, a measure of LV systolic performance, in patients with idiopathic dilated cardiomyopathy (IDC). 65 Faris and group explored the effect of gender on mortality and alcohol use, finding that men were younger and more likely than women to have a history of heavy alcohol consumption. 66 However, alcohol consumption increased the risk of death in women while showing a protective effect against death in males.
Stress
SIC, also known as Takotsubo cardiomyopathy, is characterized by acute cardiac syndrome with extensive ventricular akinesia and a distinctive LV contraction pattern. 67 Magnetic resonance imaging (MRI) reveals reduced LV ejection fraction and distinct patterns of regional ventricular ballooning. 68 SIC is highly prevalent in postmenopausal women and can be precipitated by emotional stress, neurologic injury, and various stress states such as liver transplant surgery. 69 Model organism studies suggest that circulating catecholamines initiate SIC, leading to regional akinesia. Catecholamines cause vasoconstriction of small arterioles, and their metabolites exert a direct toxic effect on the myocardium.
Diagnostic Technique
CVDs have reached epidemic levels globally 70 therefore early diagnosis is crucial for individuals exhibiting symptoms such as dyspnea, lightheadedness, abnormal heart rhythms, chest pain or discomfort, chest heaviness, and difficulty sleeping flat (Cardiomyopathy | Johns Hopkins Medicine), as it directly influences treatment outcomes and prognosis, reducing the risk of mortality. Various diagnostic techniques are available for different types of cardiomyopathies, broadly categorized as invasive and non-invasive methods.
An invasive method is generally thought of as a process that involves opening up the body on purpose by cutting it or making a puncture through the skin using medical equipment. 71 There are different invasive methods wherein first includes cardiac catheterization which is used for the detection of various heart diseases known as “Cardiac Cath” or “Heart Catheterization.” It helps a doctor to take a close look at the heart of the patient by the utilization of a catheter, a thin, flexible tube inserted inside the heart via arteries or veins. Sometimes a dye is injected to check whether the arteries are narrowed or blocked. In addition, this technique can also be used to replace valves. 72 Another important invasive method is the MyocardialBiopsywhichisused to observe whether the tissues are normal or not which is taken out using a bioptome. 73 Alongside the presence of amyloid protein levels of other substances such as iron, caesating granules, T-lymphocytes, eosinophils, and mononuclear phagocytes are also monitored.
Non-invasive methods are the primary diagnostic approach for suspected patients, including the electrocardiogram (ECG), which assesses the heart’s electrical activity. Patients suspected of cardiac disease often undergo ECG testing, where variations in ECG waves, particularly abnormal Q waves and changes in the ST-T segment, indicate the presence of disease. 74 Chest X-ray is another non-invasive technique revealing pulmonary and cardiomediastinal abnormalities. 75
Echocardiogram, which is commonly referred to as “Echo” enables us to check the overall structure and function of the heart by producing its moving pictures. It usually reveals a change in the thickness of the walls of the ventricles or the auricles and also shows any abnormality. The normal thickness of the ventricular wall is 13.6 for men and 11.2 for women. 76 Normal left atrial diameter is <4.1 in males and <3.9 in females. Size ranging from 4.1 to 4.6 for males and 3.9 to 4.2 for females is considered as mild enlargement, 4.7-5.1 for males and 4.3-4.6 for females considered as moderate enlargement, >5.2 for males and >4.7 for females s considered as severe enlargement. 77 Another important test is the Holter’s monitor which is a small medical recording device that can record the heart rhythm of a patient for about 24-72 hours. It is a type of portable ECG. It can even monitor all those small rhythms that may be missed on the ECG. 75 It shows the function of the heart on a long-term basis. An example of HCM, electrocardiographic (EKG) abnormalities, computed tomography (CT) scans, and cardiac magnetic resonance imaging (CMRI), as well as other laboratory tests, form the foundation of a correct clinical diagnosis of HCM. 35 An echocardiography, for instance, is required to determine the thickness of the walls of the heart ventricles in cases when they are not caused by exercise, aortic stenosis, or high blood pressure. This is necessary to diagnose HCM. 76
However, such diagnostic methods have some limitations which include that only the suspected patients with specific symptoms are advised for such diagnostic methods. But, what about the situation of early diagnosis without cardiomyopathy-related symptoms? Therefore, making an early diagnosis and starting therapies that will stop the disease’s course and prevent repercussions can be made easier with the use of “familial genetic testing.” This is significant, especially in light of the data showing improved long-term clinical outcomes in relatives, primarily as a result of the anticipation of diagnosis and treatment. 80
Genetic Testing in Cardiomyopathy
Recent molecular genetics research has transformed the diagnosis and treatment of cardiomyopathies like HCM by revealing their underlying pathogenesis. Collaboration among cardiologists, geneticists, and counselors is crucial for genetic testing, which identifies alterations in genes, chromosomes, or proteins to assess genetic health and pinpoint specific disease-causing genes. To be considered for clinical genetic testing, disease genes must be linked to treatment changes, with a sensitivity of at least 40%. 81 Genetic testing includes diagnostic and predictive categories, identifying disease causes and individuals at risk due to specific DNA variants. Those with mutations have a 50% chance of passing them on to offspring and require long-term surveillance. 82
Importance of Genetic Testing
Genetic testing plays a crucial role in clinical care by uncovering how genetic analysis influences disease management. Identifying mutations in family members with minimal or no symptoms allows for enhanced clinical surveillance, better medical management, and informed reproductive decisions. The primary goal of genetic testing in cardiomyopathy patients is to enable cascade testing in relatives, distinguishing those at risk from those who are not. With its ability to increase diagnostic precision and identify at-risk relatives definitively, genetic testing is invaluable for patient and family management. Different testing strategies, including whole genome sequencing (WGS), whole exome sequencing (WES), and disease-specific panels, can be adapted for patient screening and testing.83, 84
Whole Genome Sequencing
WGS is a high-throughput genomic technique used for disease gene discovery, detecting various mutations such as insertions, deletions, SNVs, copy number variations, and regulatory variants, even in the absence of relevant gene information.85, 86 Studies, like the MedSeq Project, have utilized WGS to unravel the complex genetic landscape of cardiomyopathy, identifying mutations in genes like PTPN11, ILK, and filamin-C. 87 However, limitations in variant identification methods can lead to missed detections, as seen with insufficient coverage in MYBPC3 duplications. 87 To mitigate false positives, diagnostic testing should focus on genes known to play a role in the disease, determined through methods like co-segregation analysis or case-control testing. 88 WGS in HCM families identified pathogenic variations in about 20% of cases. 89
Whole Exome Sequencing
Clinical WES has demonstrated a high diagnostic yield for rare Mendelian illnesses, providing comprehensive genomic information. Carroll et al. identified mitochondrial ribosomal protein MRPL44 alterations associated with tissue-specific symptoms like cardiomyopathy. 90 Disease-specific gene panels, coupled with Sanger sequencing, are increasingly used for monogenic disease diagnosis due to their efficiency and accuracy. 91 Seidel et al. studied 42 patients, dividing them into subgroups based on the presence or absence of DCM phenotype. They found that 22% had potentially pathogenic mutations, with genes like BAG3, DSP, LMNA, MYH7, TNNI3, TNNT2, and TTN implicated in DCM. Rare variant enrichment analysis revealed a significant accumulation of high-impact disease variants in patients with DCM phenotype. Furthermore, patients with DCM phenotype exhibited poorer event-free survival compared to those without. 92
Genotype-phenotype Correlation
Genotype-phenotype correlations elucidate physical traits or abnormalities in individuals with specific mutations. Various cardiomyopathies have been scrutinized for such correlations. For instance, diverse penetrance and sudden cardiac death rates are observed among patients with different beta-MHC gene mutations. 93 Charron and group delineated a comprehensive phenotype linked to a MyBP-C gene mutation, unveiling incomplete penetrance and a prognosis between benign and malignant alterations. 94 Anan and group investigated genotypic and phenotypic correlations in familial hypertrophic cardiomyopathy (FHC), highlighting differences in LVH patterns, age-related penetrance, and prognosis based on disease-causing genes or mutations. 95 Rai and group addressed the scarcity of data on prognosis, mutations, and genotype-phenotype correlations in Indian patients with altered MYH7 genes, revealing the phenotypic and genetic variability of cardiomyopathies in the Indian community. 4 Similarly, Loar and group explored genotype-phenotype correlations of HCM in children, adolescents, and young adults, finding higher genetic testing yields in patients diagnosed during their first two decades of life. They noted comparable phenotypes between genotype-positive and genotype-negative young patients, yet emphasized the importance of ongoing cardiac screening in genotype-positive relatives and relieving genotype-negative relatives from lifelong surveillance.96, 97
Enclosing the section, genetic screening for cardiomyopathies like HCM is pivotal for diagnosis and treatment. Collaborative testing by specialists adheres to guidelines linking disease genes to treatment changes and ensuring a minimum test sensitivity. Genetic testing serves diagnostic and predictive purposes, aiding at-risk individuals in informed management and reproductive decisions. Various strategies, including WGS, WES, and disease-specific panels, offer tailored approaches. Understanding genotype-phenotype correlations aids in personalized management, optimizing patient care and outcomes.
Treatment of Cardiomyopathy
With modern technology, understanding complex disorders like cardiomyopathy has become easier, enabling early treatment and prevention. DNA sequencing allows for genetic testing and advice, offering options like fixing mutations during in vitro fertilization. 98 Gene editing, especially for monogenic diseases like HCM, holds promise. Methods like allele-specific gene silencing have shown success in halting disease progression in animal models.99, 100 RNA interference (RNAi) has demonstrated efficacy in improving symptoms of genetic disorders such as catecholaminergic polymorphic ventricular tachycardia, HCM, and RC.101, 102 While effective for gain-of-function mutations like MYH7, its applicability to other HCM genes like MYBPC3 needs further investigation. Mearini et al. showed successful 5′ trans-splicing in HCM models, both in cardiomyocytes and in vivo in mice. 103
Machine Learning and Cardiomyopathy
Machine learning (ML) revolutionizes cardiology, offering novel approaches to grasp, diagnose, and manage cardiac conditions, especially cardiomyopathies. ML algorithms unravel complex datasets, enhancing risk prediction, disease categorization, and patient care. Studies showcase ML’s diverse applications in cardiomyopathy research, including cardiac transcriptomic data analysis for disease classification and gene identification, 104 predicting adverse events in HCM to refine risk stratification, 105 and accurately identifying high-risk heart failure patients in HCM. 106 Additionally, ML improves diagnostic accuracy, measuring LV wall thickness in HCM. 107 These findings collectively highlight ML’s pivotal role in advancing cardiomyopathy understanding and management.
Strengths and Limitations of the Review
Cardiomyopathy is considered a rare form of cardio-related disorder but results in a significant amount of mortality. Considering the strength of the present review, different aspects of cardiomyopathy have been focused which include the risk factors ranging from environmental to genetic factors with epigenetic modification also, the present review describes different types of cardiomyopathies associated with different gene mutations. Different diagnostic techniques including non-genetic and genetic testing have also been described including novel treatment strategies. Regarding the limitation of the present review, there is no specific molecule of interest described related to cardiomyopathy. Describing the cardiomyopathy disorder in a single piece of the draft is not possible due to its complexity rather it can only be characterized in terms of the tip of the iceberg.
Future Perspective
In the dynamic realm of cardiomyopathy, global health data underscores its significant burden worldwide. However, strides in epigenetics, environmental factors, genetic tests, and ML promise transformative advancements in treatment approaches. This heralds the era of precision medicine, tailoring treatment plans to individuals’ genetics, epigenetics, and environmental influences. Emphasizing early detection and prevention, new diagnostic tools and genetic testing facilitate proactive identification of at-risk individuals. Targeted treatments based on genetic insights aim to address specific mutations underlying cardiomyopathies, potentially offering cures. Concurrently, ML algorithms leverage extensive datasets to predict risks and optimize treatment strategies. Empowering patients with personalized risk assessments and lifestyle recommendations, this patient-centric approach revolutionizes healthcare engagement. Ultimately, this holistic and personalized approach has the potential to revolutionize cardiomyopathy treatment, enhance patient outcomes, and alleviate the global burden of CVD.
Conclusion
Cardiomyopathy is a disease characterized by an abundance of complexity and can be identified through the utilization of several different techniques. The condition is caused by a variety of mutations in the genes that are responsible for the development and function of the heart muscle, and environmental factors contribute to the susceptibility of the disease and increase the likelihood of developing any symptoms.
Footnotes
Abbreviations
CM, cardiomyopathy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; RCM, restrictive cardiomyopathy; AVCM, arrhythmogenic right ventricular cardiomyopathy; SNP, single nucleotide polymorphism; MyBP-C, myosin-binding protein C; FN-3, fibronectin III; LV, left ventricle; LVH, left ventricular hypertrophy; MYH7, myosin heavy chain gene; MYBPC3, cardiac myosin-binding protein C gene; MyBB-C, cardiac myosin-binding protein C; AHA, American Heart Association; BAG3, B-cell lymphoma 2 (Bcl-2)-associated anthanogene; HAND1, heart and neural crest derivatives expressed-1; POLA2, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3; PLAAT3, phospholipase A and acyltransferase 3; CCDC88B, coiled-coil domain containing 88B; LADs, lamina-associated domains; LMNA, lamin A/C; MYL2, regulatory myosin light chain; TNNT2, troponin T2; TPM1, tropomyosin alpha-1; ECG, electrocardiogram; DALYs, disability-adjusted life years; GBD, global burden disease; ARVD, arrhythmogenic right ventricular dysplasia; TTS, Takotsubo syndrome; SCN5A, sodium channel protein type 5 subunit alpha; TPM1, tropomyosin alpha-1 chain; GCAD, graft coronary artery disease; IDC, idiopathic dilated cardiomyopathy; CVDs, cardiovascular diseases; WGS, whole genome sequencing; FHC, familial hypertrophic cardiomyopathy; WES, whole exome sequencing; NGS, next-generation sequencing; TBX5, T-box transcription factor 5; TNNI3, troponin I3; FKBP5, FKBP prolyl isomerase 5.
Acknowledgments
The authors are highly thankful to the Institute of Human Genetics, University of Jammu, Department of Zoology, Lovely Professional University (Punjab) and Department of Human Genetics (Sri Pratap College, Srinagar, Cluster University Srinagar) for support in the present study.
Author Contributions
Detail of the author’s contribution, according to the CRediT (Contributor Roles Taxonomy) System: PK & SB conceptualized the study and provided supervision, SB, AS, and KM downloaded and filtered the data, SB, AS, KM, and SS drafted the manuscript, AS and SB edited the pictures, and tables, DK, NB & PK edited the manuscript and PK finalized the manuscript. All authors provided critical feedback on drafts and approved the final manuscript.
Availability of data and material
Data sharing is not available to this article as no new data were generated or analyzed in this study.
Consent for publication
Consent was not applicable, as this is a review article compiled from various research articles and guidelines and not from patients directly.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval
Ethical permission was not applicable for this article, as this is a review article drafted from various research articles and not from patients directly.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.