
Abstract
Lipid disorders play a major pathogenic role in the development of coronary artery disease, which is the major cause of mortality worldwide. In this review, the authors discuss the role of various lipid abnormalities in the causation of coronary artery disease and also highlight the ways to manage them. The roles of a healthy lifestyle and dietary patterns have been emphasized in this regard besides the significant role of drug treatment, which mostly revolves around the statin therapy.
Introduction
Among cardiovascular diseases, coronary artery disease (CAD) remains the leading cause of mortality worldwide. 1 An individual is born with a low-density lipoprotein-cholesterol (LDL-C) level of 30 mg/dL, which increases as the age advances. 2 There exists a linear relationship between the cholesterol levels and the risk of CAD, with no apparent cut-off level below which the risk declines. 2 Epidemiologic studies have shown an increased risk of nonfatal myocardial infarction (MI) and death with elevated LDL-C and total cholesterol (TC) levels. 3 Enough data suggest that cholesterol-lowering interventions are also likely to have an impact on CAD risk reduction. 3 The fact that increased cholesterol levels have a significant pathogenic role in CAD development has made lipid-lowering therapy the most important facet of medical management of this condition.
Evidence for a Causal Relationship Between Elevated Lipid Levels and CAD
The primary role of plasma lipoproteins is to transport lipids to various tissues for energy utilization, lipid deposition, steroid hormone production, and bile acid formation. 4 Lipoproteins consist of esterified and unesterified cholesterol, triglycerides, and phospholipids, and protein components named apolipoproteins that act as structural components, ligands for cellular receptors binding, and enzyme activators or inhibitors. There are 6 major lipoproteins in the blood: chylomicrons, very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), LDL, Lp(a), and high-density lipoprotein (HDL). 4 Except for HDL, all other lipoproteins have ApoB moieties, which are atherogenic. All ApoB-containing lipoproteins <70 nm in diameter [VLDL, IDL, LDL, Lp(a)], and smaller triglyceride-rich (TG-rich) chylomicrons and their remnant particles, can cross the endothelial barrier, especially in the presence of endothelial dysfunction, where they can become trapped and initiate the formation of lipid-rich atheromatous plaque.2, 4 The further growth of this atheromatous plaque depends on the duration and degree of exposure to these ApoB-containing lipoproteins in a patient’s lifetime, besides the other nonlipid factors. 4
Serum LDL-C level provides an estimate of the cholesterol mass carried by the circulating LDL particles. The latter are the most numerous ApoB-containing lipoproteins circulating in the human body. 4 Various randomized controlled trials, and epidemiological studies, have demonstrated a linear causal relationship between the plasma LDL-C and atherosclerotic cardiovascular disease (ASCVD). 3 This relationship is causal as well as cumulative and depends both on the duration of persistent increase in LDL-C and the absolute magnitude of its increase.2, 5, 6 There is enough evidence that clinical benefit is derived by therapies that lower LDL-C either by an absolute reduction in circulating LDL particles’ mass or by modification in the composition of LDL-C. The benefit in both scenarios occurs because of the absolute reduction in the ApoB levels. 6
As with LDL-C, a direct causal relationship has been noted between non-HDL-C (non-HDL-cholesterol) and the risk of CAD. Since the non-HDL-C comprises all atherogenic lipoproteins, and not just LDL, the former is proportionally about one-third more strongly associated with CAD compared to LDL-C for each standard deviation. 7 Importantly, no difference has been demonstrated between the fasting and nonfasting levels of non-HDL-C with regard to risk estimation of CAD, implicating that the lipid measurements can be simplified by using non-HDL-C (the difference between TC and HDL-C) and HDL-C levels. 8
It is a matter of debate whether TGs are causative factors of CAD; however, conflicting data suggest that they are instead the markers of risk rather than the cause, and that risk is secondary to the concomitant low levels of HDL-C, and high levels of non-HDL-C (the latter is an estimate of the total concentration of all ApoB-containing lipoproteins).9, 10 TG-rich VLDL particles and their remnants carry most of the circulating TGs. Therefore, the plasma TG concentration reflects the concentration of circulating ApoB-containing TG-rich lipoproteins. Mendelian randomization studies have suggested a causal association between plasma TGs and the risk of ASCVD; however, these studies must be interpreted with caution because nearly all the variants associated with TGs are also associated with HDL-C, LDL-C, or Lp(a). 11 The Mendelian studies strongly suggest that the causal effect of TG-rich lipoproteins and their remnants on the risk of ASCVD is determined by the circulating concentration of ApoB-containing particles, rather than by the TG content itself. 11 Therefore, until further data becomes available, the targets of treatment among individuals with elevated TG levels should be low HDL-C and high non-HDL-C rather than TGs. 10 Furthermore, in the randomized trials, medications that reduce triglyceride levels, such as extended-release niacin and fibrates, have not reduced the rates of cardiovascular events when administered in addition to appropriate medical therapy, including statins.12, 13 Before the results of the REDUCE-IT trial, 13 previous trials and meta-analyses of n = 3 fatty acid products did not show a benefit in patients receiving statin therapy.14, 15 REDUCE-IT trial demonstrated that among patients with elevated triglyceride levels despite the use of statins, the risk of ischemic events, including cardiovascular death, was significantly lower among those who received 2 g of icosapent ethyl (IPE) twice daily than among those who received placebo. 13 It is not known whether the lack of benefit from n = 3 fatty acids in previous trials may be attributable to the low dose or the low ratio of eicosapentaenoic acid (EPA) to docosahexaenoic acid (DHA).14, 15 Both the formulation (a highly purified and stable EPA ethyl ester) and dose (total daily dose of 4 g) used in REDUCE-IT were different from those in previous outcome trials of n = 3 fatty acids. However, the exact mechanism of benefit with IPE remains unclear, and whether the observed benefit was because of its triglyceride-lowering effect or because of its anti-inflammatory, antithrombotic, or membrane-stabilizing effects remains a matter of speculation. 13
HDL-C has been shown to be inversely associated with the risk of CAD, and this association persists above 70 mg/dL, albeit with some attenuation. 16 However, whether therapeutically increasing the levels of plasma HDL-C leads to any reduction in ASCVD risk, has not been conclusively demonstrated in any of the randomized trials (namely, AIM-HIGH, dal-OUTCOMES trial, ACCELERATE trial, and REVEAL trial).16-19 In the majority of these trials showing a lack of benefit, patients were already on statin therapy with optimal LDL-C goals. 20 The results further supported the notion that increasing HDL-C on its own is unlikely to be of mechanistic relevance unless coupled with an alternative pathway such as lowering LDL cholesterol. 20 Additionally, there has been increasing evidence that the quality and not the quantity of HDL is important. HDL particles promote not only reverse cholesterol transport from the periphery (mainly macrophages) to the liver but also exert pleiotropic effects on inflammation, hemostasis, and apoptosis. However, there is evidence that certain forms of “dysfunctional HDL” exist that might promote inflammation and might also persist during statin therapy and despite high HDL-C levels. 20 Whether therapies that alter the function of HDL particles will reduce the risk of ASCVD is unknown. 11
Lipoprotein(a) is a LDL particle containing an Apo(a) moiety, which is covalently bound to the ApoB component. Lp(a) can easily flux across the endothelial barrier because of an extremely small diameter size (<70 nm). 21 Elevated levels of Lp(a) augment the ASCVD risk via 2 major mechanisms: first, through a prothrombotic/antifibrinolytic effect because of the structural homology of Lp(a) with plasminogen and plasmin, and secondly, through accelerated atherogenesis resulting from the free influx and intimal deposition of Lp(a) cholesterol. 21 Enough data are suggesting a causal relationship between elevated Lp(a) levels and ASCVD 22 ; however, recent studies suggest that clinically significant reduction in the ASCVD can only be obtained by a large absolute reduction in the levels of Lp(a). This explains why earlier randomized controlled trials that evaluated therapies lowering Lp(a) by 20% to 30% (namely, niacin and CETP inhibitors) failed to demonstrate any significant reduction in the risk of adverse cardiovascular events.11, 21-23
Laboratory Measurements of Lipids and Lipoproteins
The levels of plasma lipoproteins are usually derived from their cholesterol contents, instead of measuring them directly. TC denotes 3 major classes of lipoproteins: LDL, HDL, and VLDL. Two minor lipoproteins, IDL and Lp(a), also contain smaller amounts of cholesterol. Generally, while measuring the lipid profile of a patient, the serum concentrations of TC, TG, and HDL-C are measured. Although the LDL-C can also be measured directly using enzymatic assays, it is usually indirectly derived using the Friedewald formula, as follows: LDL-C = TC – HDL-C – (TG/5) in mg/dL. For the general population, there exists a strong correlation between the derived and the direct LDL-C.11, 24 However, such calculated LDL-C has certain limitations: methodological errors and inability to use in patients with TG levels >400 mg/dL, especially in the nonfasting state. An important limitation of the Friedewald formula is the assumption of a fixed ratio of triglyceride levels to VLDL cholesterol (TG: VLDL-C) of 5:1. 24 In clinical practice, this ratio is likely to vary and may result in inaccuracies in the estimation of LDL-C, especially at high triglyceride and/or low LDL-C values. Because of this reason, many health care systems have started to endorse an alternative LDL-C estimation method (the Martin–Hopkins equation) in patients with LDL-C values below 70 mg/dL and triglycerides above 150 mg/dL. 25 Martin–Hopkins equation replaces the fixed ratio of 5 for VLDL-C estimation by using one of 180 adaptable ratios based on a patient’s individual non-HDL-C and TG values. The ratios range from 3.1 to 11.9 and are personalized to the specific lipid panel. Martin–Hopkins equation provides distinct improvements in accuracy as compared to Friedewald estimates and has an added advantage in the nonfasting setting. 25 In the postprandial state, triglyceride values may be increased, but the Martin–Hopkins equation can adapt by adjusting the estimated VLDL-C ratio. However, the accuracy of the equation also starts to decrease above a triglyceride level of 400 mg/dL. 25
The measurement of ApoB has shown superiority over the measurement of non-HDL-C and LDL-C. This is because the former is a reliable measure of the number of atherogenic particles circulating in plasma (TG-rich remnant particles, LDL, and VLDL, all contain a single ApoB molecule). The available tests measuring ApoB levels are highly accurate, standardized, inexpensive, and automated, and can be done irrespective of fasting status. 26
It is usually recommended to obtain fasting samples for lipid analyses. However, the difference which was noted as a result of fasting versus nonfasting status was small for most of the lipid parameters. For example, the level of TG increases by 27 mg/dL on average in the nonfasting state, which is unlikely to be of clinical significance for most of the individuals. Additionally, fasting versus nonfasting status carries no prognostic value for general risk screening as well. Nonetheless, in patients with diabetes mellitus, hypertriglyceridemia, and metabolic syndrome, LDL-C calculation from nonfasting parameters should be interpreted cautiously.11, 24, 25
Treatment Targets
Lipid management in CAD patients primarily targets a reduction in the LDL-C levels intending to bring down CV events.11, 27 Therefore, for patients with CAD or any form of ASCVD, it is recommended to achieve a ≥50% reduction in the level of LDL-C from the baseline and a LDL-C level of <55 mg/dL (class 1 A recommendation, ESC Guidelines 2019). 11 The target LDL-C goal (<40 mg/dL) is much more stringent for the patients experiencing a repeat cardiovascular event within the first 2 years of the previous event (regardless of the type of event) while taking the maximally tolerated and recommended lipid-lowering therapy (class 2b B).
Lifestyle Interventions
The importance of a healthy diet and lifestyle for both primary and secondary prevention of CAD cannot be underestimated. Patients should follow a dietary pattern that emphasizes the intake of fruits, vegetables, legumes, whole grains, sources of healthy protein (low-fat poultry and dairy products, nuts, and fish/seafood), and nontropical vegetable oils. Patients should limit the intake of sugar-sweetened beverages, sweets, and red meat. The intake of calories must be restricted to prevent weight gain and to ensure a loss of weight in overweight individuals. In general, it is advisable to engage in aerobic physical activity for at least 3 to 4 days/week, lasting for at least 30 min/day.11, 27
Drug Therapy
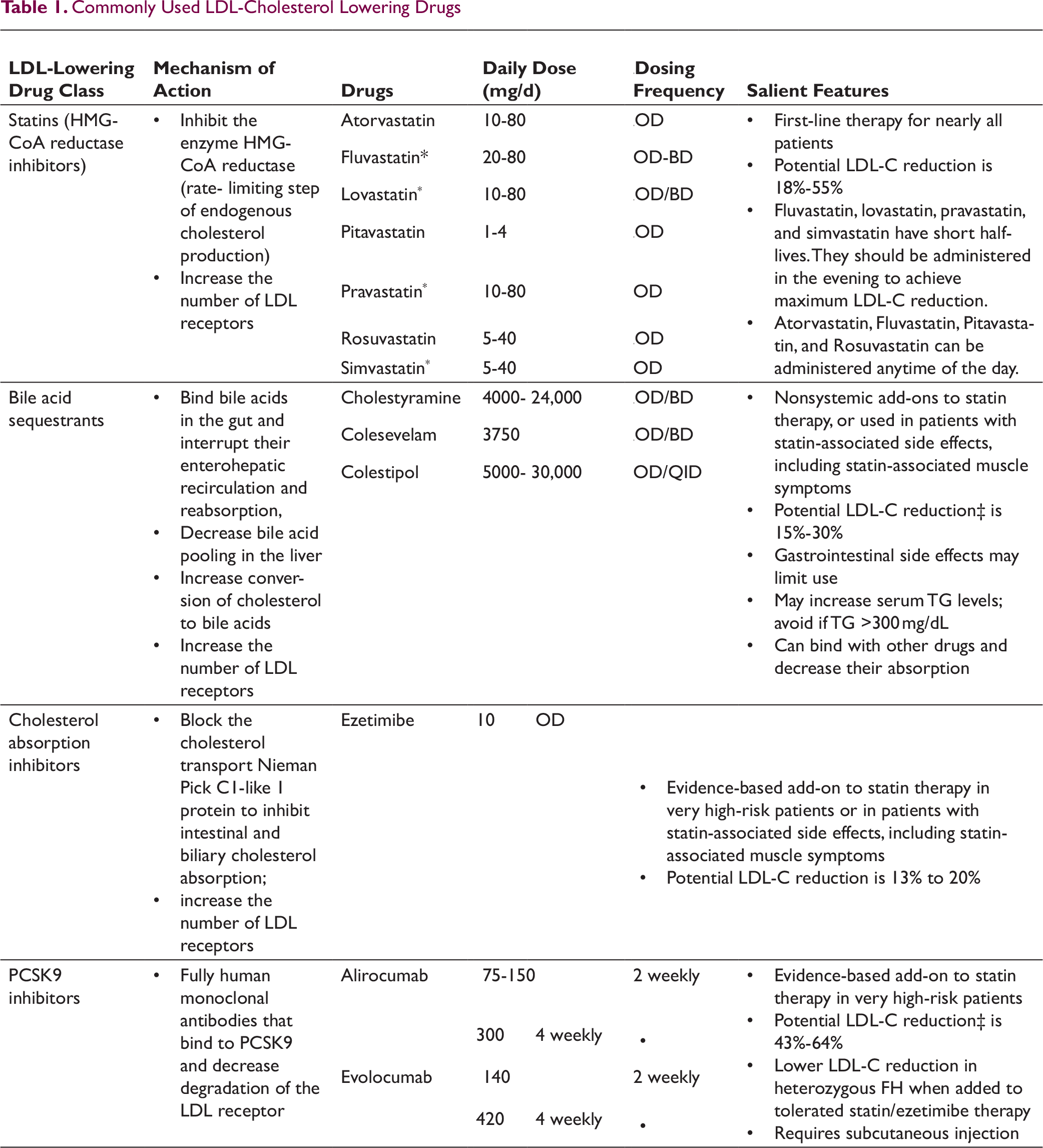
In addition to the healthy dietary pattern and healthy lifestyle interventions, drug treatment with statins forms the backbone of lipid-lowering treatment. Other drugs that lower the LDL-C include bile acid sequestrants, ezetimibe, proprotein convertase subtilisin/Kexin type 9 (PCSK9) inhibitors, inclisiran, and bempedoic acid11, 27-29 (see Table 1). Drugs lowering triglycerides namely fibrates and niacin also cause a mild reduction in LDL-C; however, randomized trials did not provide enough evidence to support their use to lower LDL-C, in addition to statin treatment. 11
Commonly Used LDL-Cholesterol Lowering Drugs
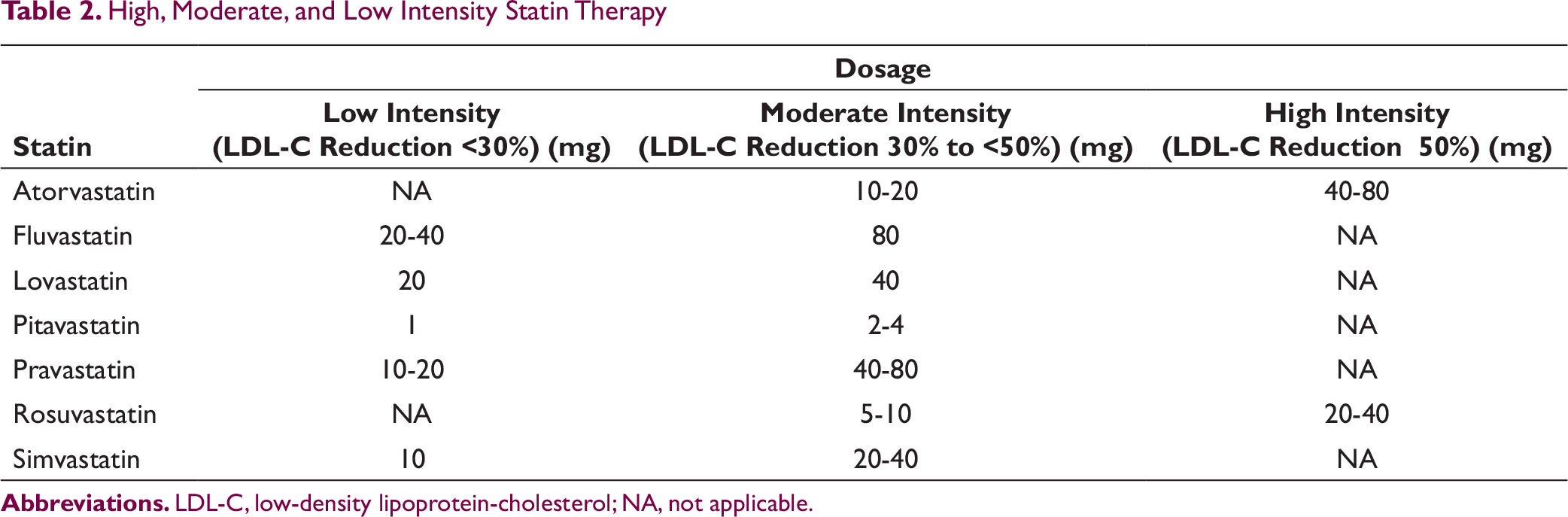
High, Moderate, and Low Intensity Statin Therapy
Early Trials, Statins for Secondary Prevention:
The first 3 trials to demonstrate the safety and efficacy of lipid-lowering treatment in secondary prevention of CV events included (a) 4S trial, (b) CARE trial, and (c) LIPID trial. The 4S (Scandinavian simvastatin survival study) trial randomized 4444 patients with angina pectoris or previous MI and serum cholesterol levels of 215 to 312 mg/dL to a lipid-lowering diet or to treatment with 20 mg/day simvastatin. 30 Over 5 years, simvastatin produced mean reductions in TC and LDL-C levels of 25% and 35%, respectively. Consequently, a 30% relative risk reduction was noted in the all-cause mortality (P <0.001). Furthermore, the CAD-related mortality was reduced by 42%, any major CAD-related event by 34%, and the need for coronary revascularization by 37%. The cholesterol and recurrent events (CARE) was a placebo-controlled trial, which also revealed similar findings, showing a significant decrease in the rates of major coronary events with pravastatin (40 mg), among patients with near-normal baseline cholesterol values. 31 The LIPID (long-term intervention with pravastatin in ischemic disease) trial (N = 9014) 32 replicated the results of 4S in providing mortality benefits with statin therapy, but that in a patient group with an overall lower total baseline cholesterol level (155 to 271 mg/dL). All the 3 trials established the safety and efficacy of statins for secondary prevention of CAD, and that too across a wide range of baseline cholesterol levels. Importantly, all these 3 trials had excluded patients with recent ACS (within the last 4-6 months before enrolment).30-32
Lipid Management in Patients With Acute Coronary Syndrome
Myocardial ischemia reduction with acute cholesterol lowering (MIRACL) was the first trial to demonstrate the benefits of early initiation of statins after ACS. 33 MIRACL was a placebo-controlled randomized trial involving 3086 patients who received 16 weeks of 80 mg/day of atorvastatin, starting 24 to 96 h after ACS. There was a significant reduction in the combined primary endpoint (recurrent symptomatic myocardial ischemia, resuscitated cardiac arrest, and nonfatal acute MI). However, certain limitations of the MIRACL trial were highlighted, which involved its short-term results, the lack of long-term safety data, and the absence of any comparator drug.
The next substantial trial involving the use of statins in ACS was PROVE-IT (the pravastatin or atorvastatin evaluation and infection therapy-thrombolysis) in MI 22 trial that compared 2 active comparators. 34 In PROVE IT-TI MI 22 trial, 4162 ACS patients (index ACS event within preceding 10 days) were randomized to either 40 mg/day of pravastatin (standard treatment) or 80 mg/day of atorvastatin (intensive treatment). Significantly lower values of the median LDL-C levels were achieved during treatment with the intensive treatment (62 mg/dL) compared to standard treatment (95 mg/dL) (P < .001). Kaplan–Meier estimates of the rates of the primary endpoint (which was a composite of all-cause death, MI, unstable angina requiring rehospitalization, revascularization, and stroke) at 2 years were 26.3% for standard therapy and 22.4% for intensive therapy, reflecting a 16% reduction in the hazard ratio in favor of intensive therapy (P<0.01). The statin-associated muscle symptoms were overall low and similar in both the treatment groups.
Furthermore, a meta-analysis was published in 2006, involving 13 randomized controlled trials, and involving 17,963 ACS patients (involving both STEMI and NSTEMI/unstable angina), demonstrating that early intensive statin therapy was beneficial for ACS patients. In this meta-analysis, the use of early statin therapy was associated with a decreased rate of mortality and CV events over 2 years of follow-up (hazard ratio, 0.81; 95% CI [0.77, 0.87]). 35 Importantly, on survival curves analysis, this benefit started to emerge between 4 and 12 months and became statistically significant by 12 months.
The first trial to demonstrate the added clinical benefit of adding a nonstatin therapy to the background statin treatment was the improved reduction of outcomes: vytorin efficacy international trial (IMPROVE-IT). This trial involved 18,134 stable patients who had a history of ACS and whose LDL cholesterol levels were within the guideline-recommended limits. 36 There was a significant reduction in the median time-weighted average LDL cholesterol level in the simvastatin–ezetimibe group (53.7 mg/dL), compared to the simvastatin-monotherapy group (69.5 mg/dL, P < .001). The Kaplan–Meier analysis showed a significant decline in the event rate for the primary endpoint at 7 years in the combination group (32.7%), compared to monotherapy group (34.7%) (absolute risk reduction 2%; hazard ratio, 0.936; 95% CI [0.89, 0.99]; P <0.02). No significant difference was noted in the side effect profile (including muscle-related, gallbladder, hepatic adverse effects, and cancer). The clinical benefit of the addition of ezetimibe to statin therapy was augmented in patients with prior CABG (n = 1684 out of 18,134). 37 The absolute risk reduction in the primary endpoint, as a result of ezetimibe addition, was higher in the prior CABG patients (8.8%; 95% CI [3.1,14.6]), compared to those without prior CABG (1.3%; 95% CI [0,2.6]) (P-interaction = 0.02).
Among patients with ACS, it is recommended to obtain a lipid profile as soon as possible after admission, because LDL-C levels tend to decrease during the first days of ACS. Patients do not have to be fasting, as this has little impact on the LDL-C levels.11, 27 Current recommendations suggest routine and early use of high-intensity statin therapy in patients with ACS.11, 27 An additional advantage of early high-intensity statin therapy in ACS patients undergoing early invasive treatment is protection from contrast-induced nephropathy. 38 Importantly, the lipid-lowering therapy should be continued indefinitely, without reducing the dose in patients who are tolerating the treatment, regardless of how low the LDL-C falls. The treatment goal is to reach a 50% LDL-C reduction from baseline and an LDL-C goal of <1.4 mmol/L (<55 mg/dL). In those with recurrent events within 2 years while taking maximally tolerated statin therapy, a goal of <1.0 mmol/L (<40 mg/dL) for LDL-C should be considered. 11 Lipid levels should be re-evaluated 4 to 6 weeks after ACS to determine whether treatment goals have been achieved and to check for any safety issues; the therapeutic regimen can then be adapted accordingly. The use of lower intensity statin therapy should be considered in patients at increased risk of adverse effects with high-intensity statin therapy, such as in the elderly, patients diagnosed with hepatic or renal impairment, or in the case of a potential risk of drug–drug interactions with other essential concomitant therapies. Nonetheless, if the target goals are not achieved or if statin intolerance occurs, Ezetimibe and PCSK9 inhibitors can be considered sequentially. 11 Importantly, PCSK9 inhibitor can be considered early after ACS (during hospitalization, if possible), in patients who present with an ACS and whose LDL-C levels are not at goal, despite already taking a maximally tolerated statin dose and ezetimibe. 11
Lipid Management in Patients With Chronic Stable Angina
The landmark results from PROVE IT trial laid the foundation for all major guidelines recommending a LDL-C goal of <70 mg/dL in high-risk patients.11, 27 However, it was yet to be seen whether the safety and efficacy of long-term intensive statin treatment documented in ACS patients could be replicated in stable CAD patients as well. This question was addressed by treating to new targets (TNT) 39 and the incremental decrease in endpoints through aggressive lipid lowering (IDEAL) trials. 40 In the TNT trial, 10,001 CAD patients from 14 countries were enrolled. All these patients had baseline LDL levels < 130 mg/dL and were administered 10 mg/day or 80 mg/day of atorvastatin in a double-blind, parallel-group design. The median follow-up duration was 4.9 years. Mean LDL-C levels were reduced to 77 mg/dL in the 80 mg group compared to 101 mg/dL in the 10 mg group. The primary endpoint, a composite of cardiovascular mortality, MI, resuscitated cardiac arrest, and stroke, occurred in 10.9% of patients in the 10 mg and 8.7% of patients in the 80 mg group (P = .0002). The benefit with 80 mg atorvastatin was much more pronounced in patients with prior CABG, diabetes mellitus, metabolic syndrome, and chronic kidney disease. The risk of hospitalization for heart failure and stroke was also significantly lower in the 80 mg group. However, no significant intergroup difference was observed in the overall mortality. Besides a higher incidence of persistent liver transaminitis with 80 mg atorvastatin (P < .001), no other significant difference in the adverse events was recorded among the 2 groups. 39
The results of the IDEAL trial involving 8888 patients and >20,000 patient-years of follow-up, further established the safety and benefits of intensive statin therapy in stable CAD management. 40 Based on all this evidence, guidelines endorsed by all major societies recommend high-intensity statins in all patients with established CAD, especially in those <75 years of age.11, 27
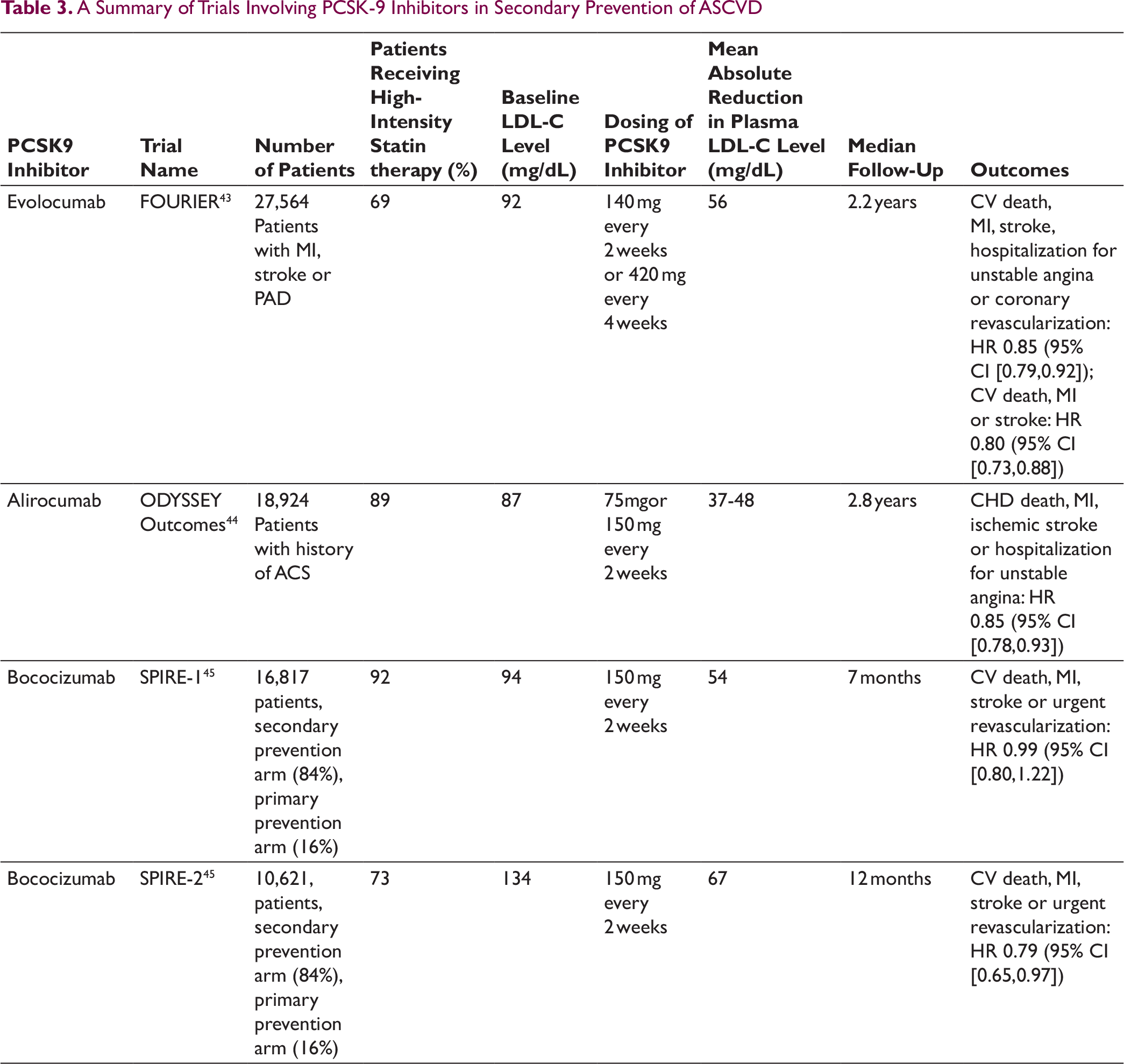
A Summary of Trials Involving PCSK-9 Inhibitors in Secondary Prevention of ASCVD
Novel Nonstatin Therapy to Reduce LDL-Cholesterol
Recent years have witnessed multiple cardiovascular outcome trials showing the excellent efficacy of PCSK9 inhibitors in reducing the risk of major vascular events. 42 This benefit with PCSK9 inhibitors persisted in patients with a baseline LDL-C level less than 70 mg/dL, in whom the level reduced to approximately 20 mg/dL, which is well below the current guideline-recommended targets. In a randomized placebo-controlled trial of PCSK9 inhibitor evolocumab, involving 27,564 patients with established ASCVD, a 15% reduction was achieved in the composite of CV death, MI, stroke, hospitalization for unstable angina, or coronary revascularization (3-year rate, 12.6% vs 14.6%, P < .001). 43 A summary of trials involving PCSK9 inhibitors in secondary prevention of ASCVD is listed in Table 3. Inclisiran is a small interfering RNA (siRNA) that reduces hepatic PCSK9 synthesis.28, 46 In the ORION 11 trial, inclisiran reduced LDL-C by over 50% in the patients with stable ASCVD or ASCVD-risk equivalent (type 2 diabetes, familial hypercholesterolemia, ≥20% ASCVD risk) and elevated LDL-C despite maximum tolerated statin therapy. 47 A noteworthy advantage of inclisiran is its 6-monthly dosage regimen, which has the potential to increase treatment adherence. Besides injection-site adverse events, no other significant adverse effects have been reported.46, 47 The post hoc analysis of ORION 11, based on discharge summaries and adverse event reports, demonstrated a lower incidence of cardiovascular events among patients randomized to inclisiran compared to placebo.47, 48 Cardiovascular outcomes of inclisiran are currently being assessed in a dedicated outcomes trial (ORION-4). 48
Bempedoic acid, a novel agent, is an oral, once-daily, nonstatin LDL cholesterol-lowering drug that inhibits ATP citrate lyase, a key enzyme linking glucose catabolism to lipogenesis by catalyzing the formation of acetyl-CoA from mitochondrial-derived citrate for de novo synthesis of fatty acids and cholesterol. 29 Based on the results of recent clinical trials, bempedoic acid was approved for use by the US Food and Drug Administration in February 2020 for the treatment of adults with heterozygous familial hypercholesterolemia or established ASCVD, who require additional lowering of LDL-C. 49
Conclusion
Statin therapy has been shown to be effective in all CAD patients. Intensive treatment with statins retards the progression of atherosclerotic disease and mitigates long-term risk of cardiovascular events in CAD patients. No threshold LDL-C level has been elucidated, below which the beneficial effects of intensive statin therapy disappear. The concept of “lower the better” has been advocated by a few authors to control LDL-C levels among patients with CAD. Besides causing a reduction in the LDL-C levels, intensive statin therapy also reduces inflammation, which is perhaps responsible for its beneficial effect among ACS, especially when started early. Beyond statin therapy, there are various other drugs that favorably alter lipid profiles, but long-term data are limited and still await validation in large-scale outcome studies. Lastly, the importance of a healthy lifestyle and dietary pattern is often neglected in lipid management among CAD patients, and should always be encouraged.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.