
Abstract
Rett syndrome (RTT) is a rare X-linked syndrome that predominantly affects girls. It is characterized by a severe and progressive neurodevelopmental disorder with neurological regression and autism spectrum features. The Rett syndrome is associated with a broad phenotypic spectrum. It ranges from a classical Rett syndrome defined by well-established criteria to atypical cases with symptoms similar to other syndromes, such as Angelman syndrome. The first case of a Moroccan female child carrying a R306X mutation in the MECP2 (Methyl-CpG-Binding Protein 2) gene, with an unusual manifestation of Rett syndrome, is presented here. She showed autistic regression, behavioral stagnation, epilepsy, unmotivated laughter, and craniofacial dysmorphia. Whole exome sequencing revealed a nonsense mutation (R306X), resulting in a truncated, nonfunctional MECP2 protein. The overlapping phenotypic spectrums between Rett and Angelman syndromes have been described, and an interaction between the MECP2 gene and the UBE3A (Ubiquitin Protein Ligase E3A) gene pathways is possible but has not yet been proven. An extensive genetic analysis is highly recommended in atypical cases to ensure an accurate diagnosis and to improve patient management and genetic counseling.
Introduction
Rett Syndrome is an X-linked neurodevelopmental disorder 1 that predominantly affects girls with a worldwide prevalence of 1/10 000 to 15 000. 2 The Moroccan Association of Rett syndrome reported 20 cases annually. 3 The typical phenotype of Rett syndrome is characterized by normal development for the first 6 to 18 months after birth, followed by 4 stages of disease progression. The first stage is characterized by developmental and postural delays, hypotonia, and the beginning of the microcephaly. This stage lasts from some weeks to months. The second stage includes a loss of previously gained motor skills and communication, the appearance of stereotypic and repetitive hand movements, and breathing irregularities. The microcephaly becomes more obvious and seizures may occur. The average period of this stage is weeks to months and up to 1 year. Stage 3 is a plateau period. During this stage, the patient can show signs of recovery. Motor deterioration characterizes the last stage, patients suffer from a severe physical disability, wasting, dystonia, bradykinesia, and scoliosis, with a possibility of being wheelchair dependent. Besides this typical phenotype, we note the presence of a spectrum of phenotypes called “atypical Rett syndrome.”
Several genes have been linked to typical and atypical Rett syndrome. Indeed, mutations in the MECP2 gene explain 95% of typical Rett syndrome cases and 75% of atypical Rett syndrome cases. 4 Other genes have been also found to be implicated in Rett syndrome such as CDKL5, FOXG1, STXBP1, TCF4, SCN2A, WDR45, and MEF2C. On the other side, some genes have been linked to other syndromes namely UBE3A in Angelman syndrome, SHANK3 in Phelan–McDermid syndrome, or TCF4, CNTNAP2, and NRXN1 genes in Pitt–Hopkins syndrome etc. 5
In this report, we present the first Moroccan child with atypical Rett syndrome carrying a MECP2 mutation R306X.
Case Presentation
We report the case of a 13-year-old Moroccan female patient. She is the second child of 3 siblings of non-consanguineous parents. The patient’s mother had a history of one uninvestigated miscarriage. Her pregnancy was normal, carried to term with normal vaginal delivery and without neonatal suffering. Family investigation revealed the absence of similar cases within the family (Figure 1).
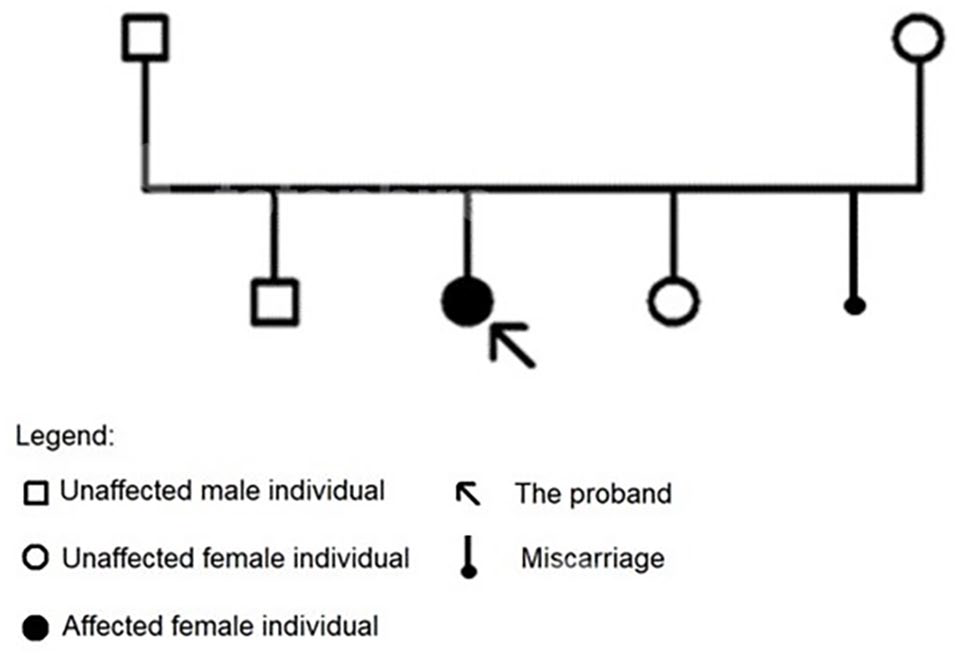
Genealogical tree of the proband.
The clinical manifestation comprises psychomotor delay (walking at 24 months), language delay (her speech includes only a few words), profound intellectual disability, with autistic regression and behavioral stagnation, epilepsy since she was 6 months old, unmotivated laughter, craniofacial dysmorphia characterized by a receding forehead, an elongated face, a broad nose, hypotrophy of the extremities, nocturnal strabismus, and manual stereotypies. Her secondary sexual characteristics development was normal. The initial electroencephalogram (EEG) performed at the age of 2 years reported a disturbed background rhythm made of alpha waves at 5 cycles per second mixed with theta waves, with the presence of slow waves and sometimes diffuse symmetrical and synchronous waves at the end of the tracing, the very slow tracing was in favor of diffuse cerebral suffering. A later EEG performed at 3-year-old was normal. The cerebral MRI didn’t show any abnormalities. The blood screening, hormonal and other radiological investigations were all normal.
Initially, we suspected an Angelman syndrome because of the presence of several signs, such as intellectual disability, microcephaly, speech delay, seizures, stereotyped movement, unmotivated laughter, and autistic behavior. Thereby, the initial genetic screening was without abnormalities: Constitutional Standard Karyotype (46, XX), array CGH, and analysis of methylation at the SNRPN locus by MS-PCR (test for Angelman syndrome). However, the Whole Exome Sequencing has identified a heterozygous pathogenic variant in the MECP2 gene NM_001110792.1: c.916C>T (p.Arg306*), which confirmed the diagnosis of Rett Syndrome.
Discussion
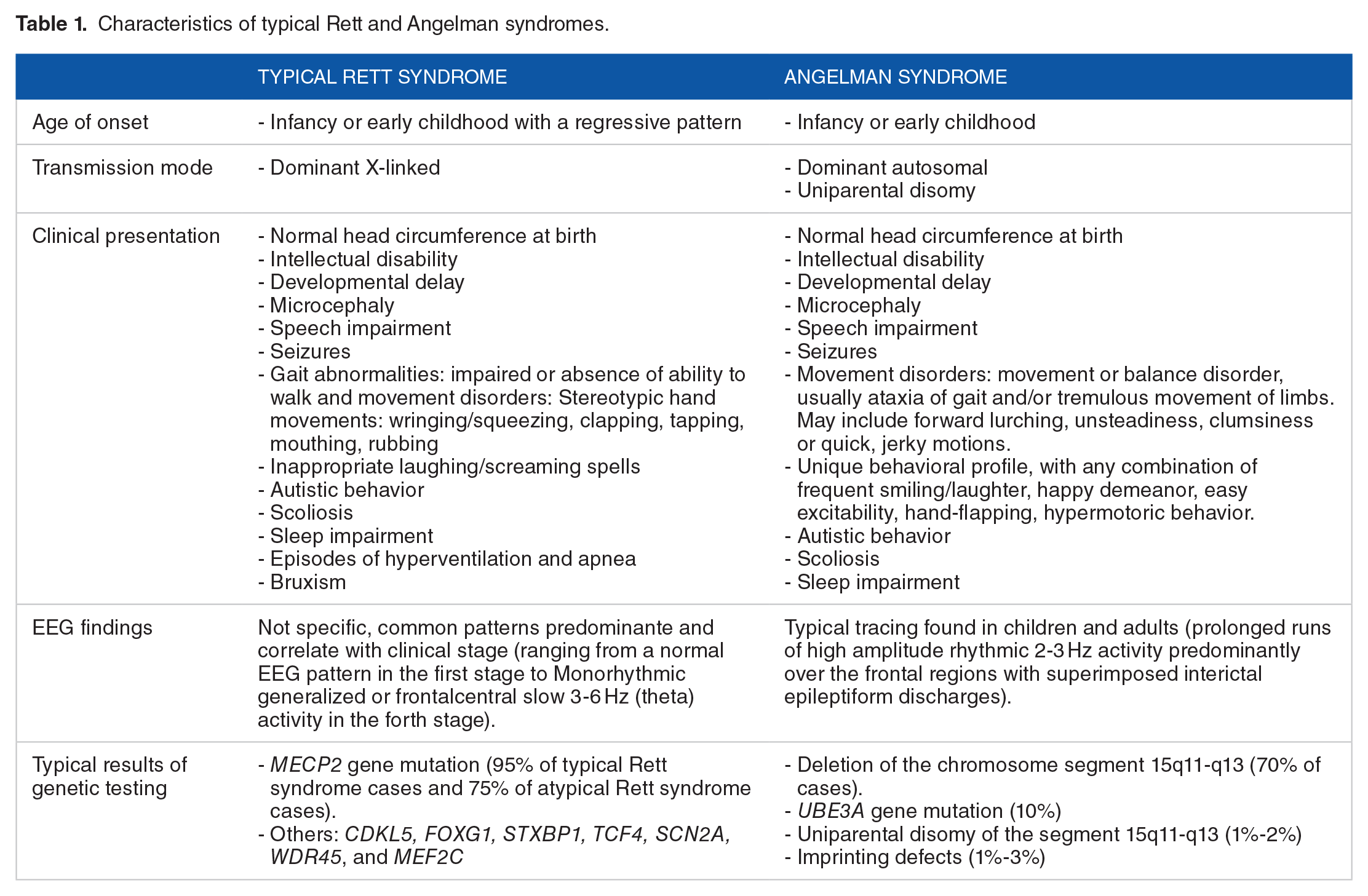
Rett and Angelman syndromes share some common symptoms, such as intellectual disability, microcephaly, speech impairment, seizures, movement disorders, and autistic behavior (Table 1). 6 These similarities can lead to missed or delayed diagnosis. The classic Rett syndrome diagnostic criteria were defined initially in 1988 and revised in 2010. The latest version summarizes criteria for the diagnosis of typical and atypical Rett syndrome (Table 2). In the ‘90s, the atypical Rett diagnostic criteria were introduced to include children who do not meet all the typical Rett diagnostic criteria. On the other hand, the Angelman syndrome is a neurodevelopmental disorder with several symptoms: a severe developmental delay, speech disorder with no or minimal use of words, high nonverbal and receptive communication skills, movement disorders like ataxic and/or limb tremulous movements, unique behavioral (frequent and inappropriate laughter, easily excitable personality, hand flapping movements and short attention span), delayed and disproportionate growth in head circumference leading to microcephaly by the age of 2 years, early onset seizures (<3 years of age), a characteristic EEG pattern with large amplitude slow spike waves and triphasic waves. 7 Among the clinically diagnosed cases of Angelman syndrome, 90% present a positive molecular test, while the remaining negative cases are called Angelman-like Syndrome. These similarities between both syndromes raise a rather intriguing question: does the MECP2 gene responsible for Rett syndrome and the UBE3A gene implicated in Angelman syndrome regulate each other or have a common physiological pathway?
Characteristics of typical Rett and Angelman syndromes.
Diagnosis criteria for typical and atypical Rett syndrome*.

*Adapted from: Rett Syndrome Diagnosis. International Rett Syndrome Foundation. Retrieved December 13, 2021, from https://www.rettsyndrome.org/about-rett-syndrome/rett-syndrome-diagnosis/.
The MECP2 is a gene located on Xq28. 8 The inheritance of a mutation in the MECP2 gene can manifest in an X-linked dominant model that results in Rett syndrome or an X-linked recessive dominant model observed in other diseases such as encephalopathy, X-linked Lubs-type syndromic intellectual developmental disorder, and other forms of non-specific mental retardation. 9 About 99% of Rett syndrome cases are sporadic predominantly of paternal origin, 10 however, familial cases of maternal origin are rarer. 11 In addition, the MECP2 gene is subject to X inactivation. Hence, the variation in Rett phenotypes and the appearance of atypical forms are due to the ratio of inactivated X/activated X 12 which makes the correlation between genotype and phenotype inconsistent therefore the females with favorably skewed X-chromosome inactivation (XCI) may have mild or no pathogenic phenotype.
The MECP2 gene comprises 4 exons encoding for the methyl-CpG binding protein 2, a nuclear protein, present in 2 distinct isoforms MECP2A and MECP2B 13 that play several roles. Indeed, it’s an important epigenetic modulator expressed mainly in the brain. It controls gene expression by binding to methylated DNA and histones. 14 In addition, MECP2 has been found to act as a transcriptional repressor and activator. 15 MECP2 deficiency may cause up or downregulation of specific genes (BDNF, GAMT, DLX5, DLX6, FKBP5, SGK1, FXYD1, MPP1 are upregulated, and UBE3A and GRID are downregulated). 16
MECP2 protein is characterized by functional domains (Figure 2) namely the methyl-CpG binding domain (MBD) that recognizes and binds to methylated CpGs sites, especially to AT-rich regions. It also binds to unmethylated DNA with lower affinity, suggesting that MBD may play another role than DNA inactivation. 17 The second domain is the transcriptional repression domain (TRD) that recruits chromatin remodelers and gene repressors, such as histone deacetylase (HDACs) and Sin3a. 18 The nuclear localization signal (NLS) is another domain located within the TRD domain, which is implicated in the nuclear localization of MECP2. However, it was reported that NLS is not a detriment for the nuclear localization because MBD is sufficient and mutation in this sequence doesn’t induce Rett syndrome. 19 MECP2 contains also an N-terminal domain (NTD) and an intervening domain (ID). This latter increases the structural stability of MBD and provides an additional site involved in chromatin remodeling and looping. It interacts with nucleosomes by replacing histone H1. 20 The C-terminal portion includes 2 DNA binding regions (CTD α and β) 21 involved chromatin structure regulation by mediating nucleosomal array compaction and oligomerization. 22 MECP2 pathogenic mutations predominantly occur in the MBD (such as R106W, R133C et T158M), the ID (R168X) and TRD sites (R255X, R270X, R294X and R306C) highlighting the importance of these domains in the protein’s function 23 (Figure 2).

MECP2 gene structure showing the localization of the nonsense mutation R306X, and the most common pathogenic mutations of Rett syndrome.
Our patient presents a nonsense mutation (R306X) in the TRD2 domain (Figure 2),24,25 This mutation leads to premature termination of the protein that may cause structural changes affecting the activity and the stability of the protein. 22 Thus, the truncated protein contains the NTD-MBD-ID-NLS domains, while the TRD domain is truncated and the CTD domain is eliminated.
A study of the R168X nonsense MECP2 mutation that produces a truncated protein consisting only of NTD-MBD domains has demonstrated that the abnormal protein can bind to nucleosomal arrays but cannot generate a higher-order chromatin structure. This finding suggests that the MBD cannot mediate chromatin condensation on its own. 26 Shahbazian et al 27 have investigated the R308X nonsense mutation that results in an elimination of the CTD domain and causes Rett syndrome in mice. The study revealed that the absence of the CTD domain causes hyperacetylation of the histone H3 and could be responsible for alteration in chromatin structure. 27 Moreover, the R306C missense mutation induces a loss of MECP2 protein interaction with NCoR/SMRT responsible for the Rett syndrome. 25 Thereby, the R306X nonsense mutation might have the same consequence or be more severe because of the additional loss of the CTD site.
On the other side, the UBE3A gene, located in chromosome 15q11.2, is a maternally expressed imprinted gene. 28 Loss of its expression is responsible for Angelman syndrome. 29 This gene encodes the ubiquitin-protein ligase E3A an essential protein in the ubiquitin-proteasome pathway. This latter is important for maintaining synaptic plasticity and normal neuronal activity. Indeed, the E3A promotes proteins degradation via the ubiquitin-proteasome pathway such as p53, p27, Arc, and ephexin5.29 -31 Consequently, the loss of UBE3A function causes perturbations in the levels of these proteins in the neuronal cell.
The relationship between MECP2 deficiency and UBE3A expression is still highly controversial. Samaco et al 32 have found that MECP2 causes UBE3A dysregulation. Indeed, they observed lower levels of UBE3A/E6AP expression in MECP2 knockout mice strains and cerebral human postmortem samples affected by Rett, Angelman, and autism disorders. Moreover, the MECP2 deficiency doesn’t alter UBE3A gene imprinting, which indicates that MECP2 may affect the UBE3A in other more complex ways, like chromatin loop formation previously studied in the imprinted Igf2/H19 region. 33 Makedonski et al 34 confirmed these findings and showed the epigenetic abnormalities at the PWS imprinting center caused by MECP2 deficiency. Indeed, MECP2 gene loss alters histone modifications (elevation in histone H3 acetylation and H3 (K4) methylation and reduction in H3 (K9) methylation at the imprinting center). It results in the loss of imprinting of the UBE3A antisense gene in the brain, a rise in UBE3A antisense RNA levels, and a decrease in UBE3A synthesis. In the same perspective, Kim et al 35 have suggested that MECP2 and E6AP share transcriptional control over the expression of mutual target genes, they discovered that decreased UBE3A expression reduces the pathogenic phenotype in drosophila due to MECP2 overexpression, and direct physical interaction between E6AP and MECP2. Furthermore, they concluded that these 2 genes do not modulate each other’s expression in contrast with earlier researches.36,37 Thus, the overlapping spectrum of Rett and Angelman syndromes can be clarified by the relationship between MECP2 and UBE3A genes.
Clinically, few molecular studies have focused on MECP2 mutation identification in Angleman-like phenotypes. Psoni et al have detected a c-terminal in-frame 6 bp deletion (c.1159_1164del6) in a female patient with Angelman-like syndrome. 38 In another case report, Kharrat et al have described an Angelman-like phenotype with a double mutation in the MECP2 gene namely c.397C > T in trans and c.608C > T. 39 In addition, Milani et al have reported a case study of a patient with an atypical Rett syndrome (Angelman-like) phenotype with a missense T158M mutation in the MECP2 gene. 40
In conclusion, more studies are needed to explore the association between the MECP2 and UBE3A pathways in order to explain the overlapping phenotype. Furthermore, genetic testing is recommended for atypical cases to improve diagnosis and patient management, and genetic counseling.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
WB and HD and TA conceived of the presented idea. SB collected the patient’s Data. WB researched the topic and wrote the manuscript. TA verified the analytical methods and encouraged WB to investigate the mutation position and its effect on the protein. HD and BG supervised the findings of this work. All authors discussed the results and contributed to the final manuscript.
Ethical Statement
This material is the authors’ own original work, which has not been previously published elsewhere. The paper properly credits the meaningful contributions of co-authors. The informed consent was obtained from the legal representative of the patient.