
Abstract
The care of transgender individuals is multidisciplinary including medical, surgical, psychiatric, and psychological health professionals. We here report 2 cases of nonclassic congenital adrenal hyperplasia (CAH) coincidently discovered during the clinical and biochemical evaluation prior to hormonal treatment for gender dysphoria. Elevated 17-OH-progesterone and androstenedione led to further investigations including adrenocorticotropic hormone stimulation test and genetic evaluation of 21CYPA2 confirming the diagnosis of CAH in both individuals. The findings may have implications for future surgical treatment and in the case of transwomen for the choice of antiandrogenic treatment. These cases therefore confirm the importance of thorough clinical and biochemical evaluation of transgender individuals before initiation of hormone therapy.
Introduction
Gender dysphoria (GD) is defined as clinically significant distress resulting from an incongruence between the experienced gender and the gender assigned at birth. 1 Diagnostic criteria for GD were revised in DSM-5 and includes a subtype of patients with a disorder of sex development (eg, sex chromosome disorders or congenital adrenal hyperplasia [CAH]). 1
The care of transgender individuals is multidisciplinary including medical, surgical, psychiatric, and psychological health professionals. Clinical practice guidelines concerning the care of patients with GD have been published and continuously reviewed by the Endocrine Society 2 and the World Association for Transgender Health 3 and include recommendations about medical and psychiatric evaluation prior to initiation of hormone treatment.
In this report, we describe 2 cases of nonclassic CAH coincidently discovered during the clinical and biochemical evaluation prior to hormonal treatment for GD.
Methods and Materials
During the period from January 2016 to December 2020, a total of 241 patients (male to female [MtF] n = 63 and female to male [FtM] n = 178) below the age of 19 years were referred to the Department of Growth and Reproduction, Rigshospitalet, Denmark, the national pediatric center for GD, for hormone therapy.
Prior to referral for hormone therapy, a full physical examination and screening for any psychopathology was carried out at the Child and Adolescent Mental Health Center, Bispebjerg Hospital. In at least 5 face-to-face interviews between the patient, the parents, and a mental health professional at the Sexological Clinic, Rigshospitalet, the gender and body dysphoria were evaluated. The final referral for hormonal therapy was discussed and decided at a joint conference of the multidisciplinary team including representatives from all departments. The treatment and related consultations were carried out in agreement with international guidelines.2,3
Standard Evaluation
All patients underwent a standardized medical evaluation including blood sampling and whole-body DXA-scan prior to treatment.
Blood samples were analyzed for liver and kidney function, thyroid hormones, lipids, vitamin D, hematology, sex hormones, and karyotype. This included measurement of serum concentrations of luteinizing hormone (LH), follicle stimulating hormone (FSH), androgens (testosterone [T], androstenedione, dehydroepiandrosterone-sulfate [DHEAS], 17-OH-progesterone [17-OH-P]), and estrogens (estradiol [E2], calculated free estradiol, estrone, and calculated free estrone). LH and FSH were measured by a time-resolved immunofluorometric assay (DELFIA; Wallac Inc, Turku, Finland), steroid hormones were measured by Isotope-dilution TurboFlow-LC-MS/MS.
Supplementary Evaluation
In both cases, ACTH-stimulation test was performed to evaluate the hypothalamic-pituitary-adrenal axis. Blood samples were drawn at baseline, 30 and 60 min after the administration of 250 µg ACTH (Synacthen; Novartis Healthcare, Copenhagen, Denmark) intravenously. At each time point, the concentration of cortisol and 17-OH-P was measured by competitive electrochemiluminescence-immunoassay (Cobas 8000; Roche Diagnostics, Hvidovre, Denmark) and isotope-dilution TurboFlow-LC-MS/MS, respectively. ACTH was measured at baseline by a sandwich chemiluminescent immunoassay (IMMULITE 2000 XPi; Siemens Healthcare, Ballerup, Denmark).
Genetic Analysis
In both cases, a genetic analysis was performed by polymerase chain reaction and direct sequencing of the coding and flanking exons in the CYP21A2 gene except part of exon 3.
Case 1
A 17-year-old FtM transgender individual was referred for hormone therapy. The patient presented with gender and body dysphoria in adolescence and did not experience a clear gender incongruence in childhood although a vague attraction toward the opposite gender stereotype was reported. Clinical examination revealed normal height (156.3 cm), weight (55.6 kg), body mass index (BMI) (22.8 kg/m2), and blood pressure (120/71 mm Hg). Pubertal development was assessed to Tanner breast stage B5 with a male pattern of body hair distribution (chest, face, lower back, and extensive hair growth on the legs). The karyotype was 46,XX.
Medical history included evaluation of early pubarche at the age of 7 years. This was not investigated further. Menarche occurred at the age of 11 years. Nothing further was noted in terms of any history of medical or psychiatric comorbidities and the patient received no medication.
Blood samples before hormone treatment showed elevated basal 17-OH-P, T, and androstenedione, but normal DHEAS (Figure 1). An ACTH stimulation test showed a normal increase in cortisol (baseline: 504 nmol/L, 30 min: 553 nmol/L, and 60 min: 555 nmol/l), normal basal ACTH (18 pmol/L), and elevated 17-OH-P concentrations (baseline: 54 nmol/L, 30 min: >60 nmol/L, and 60 min: >60 nmol/L) at all time points. The patient was homozygous for the CYP21A2-mutation V281L, which confirmed the diagnosis of nonclassic CAH.
Concentrations of Testosterone, 17-OH-P, DHEAS, and Androstenedione in an FtM, Case 1, Before Initiation of Hormone Treatment for Gender Dysphoria
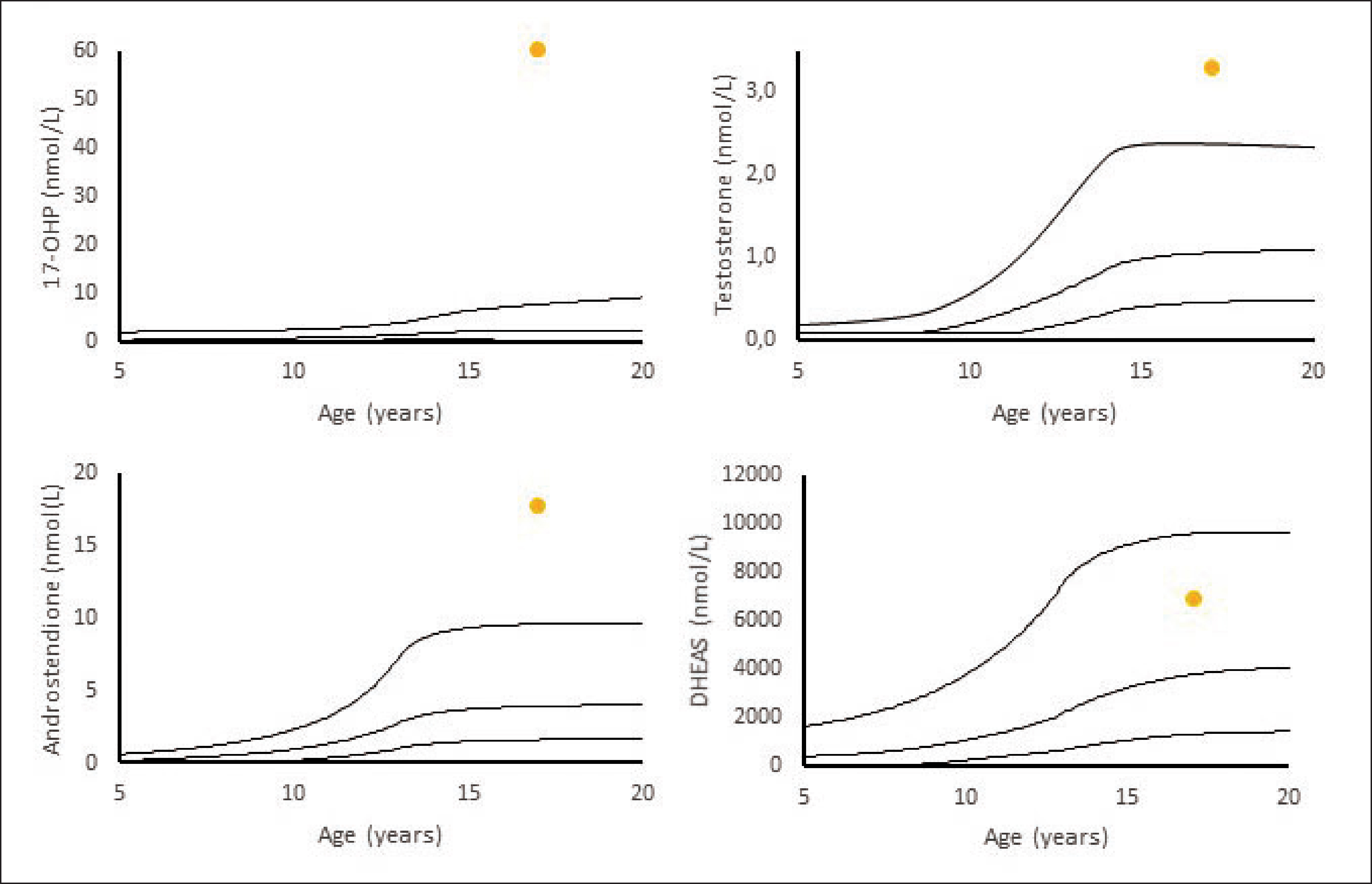
Case 2
A 16-year-old MtF transgender individual was referred for hormone therapy. The patient presented with gender and body dysphoria in adolescence and did not experience a clear gender incongruence in childhood although a vague attraction toward the opposite gender stereotype was reported. The physical examination included measurements of height (185.0 cm), weight (68.8 kg), BMI (20.1 kg/m2), and blood pressure (130/86 mm Hg). The patient reported early pubarche. There was no significant past medical history or psychiatric comorbidities and the patient received no medication. The karyotype was 46,XY.
Initial blood samples showed elevated 17-OH-P and androstenedione, but normal T and DHEAS (Figure 2). An ACTH-stimulation-test showed normal response in cortisol (baseline: 586 nmol/L, 30 min: 671 nmol/L, and 60 min: 694 nmol/L), and basal ACTH (10 pmol/L), but elevated concentrations of 17-OH-P at all time points (baseline: 22,4 nmol/L, 30 min: >60 nmol/L, and 60 min: >60 nmol/L).
Concentrations of Testosterone, 17-OH-P, DHEAS, and Androstenedione in an MtF, Case 2, Before Initiation of Hormone Treatment for Gender Dysphoria
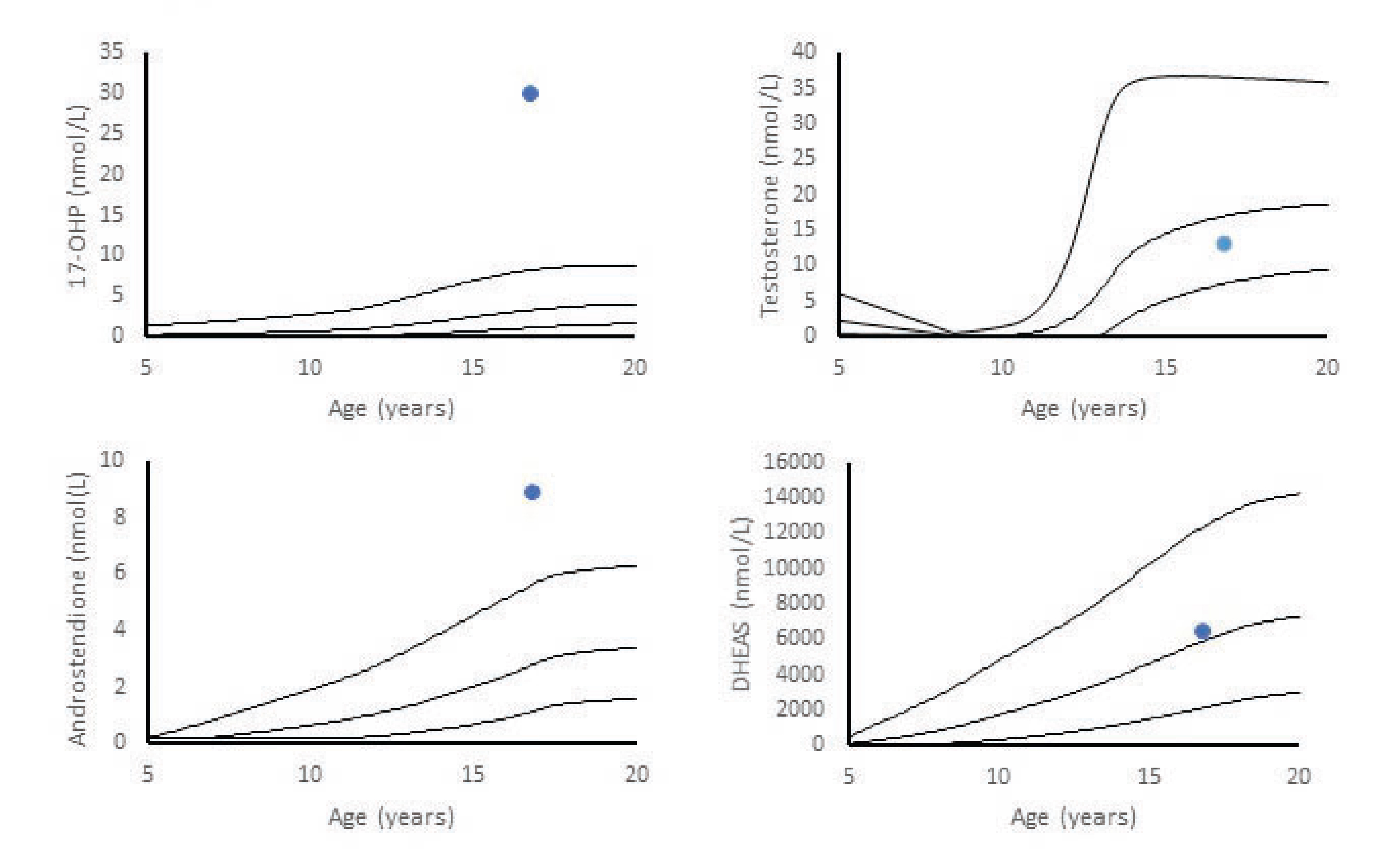
The patient was compound heterozygous for two mutations (c.1360C>T and c.1451_1452delinsC) in CYP21A2 confirming the diagnosis of nonclassic CAH.
Discussion
We here report 2 cases of nonclassic CAH coincidently discovered during the standard endocrine investigations prior to treatment for GD in a FtM and MtF individual. None of the patients defined themselves in any gender-nonconforming category before coming out as transgender and seeking gender reassignment. As such, these patients did not present any essential differences from other transgender children and adolescents referred to our treatment program. Both had a history of early pubarche, but this was not evaluated further. Elevated 17-OH-P led to further investigations including ACTH stimulation test and genetic evaluation of 21CYPA2 confirming the diagnosis of CAH in both individuals. We do not believe that the CAH in our patients was causally related to being transgender.
These findings did not change our treatment protocol with GnRH analogues and gender affirming hormones. According to international guidelines, treatment with daily hydrocortisone in asymptomatic nonclassic CAH is not recommended. 4 However, according to guidelines, both individuals were advised to take stress doses of glucocorticoids during illness and future surgery. The transwoman may over time require additional antiandrogenic therapy to further reduce unwanted symptomatic levels of androgens. These cases therefore confirm the importance of thorough clinical and biochemical evaluation of transgender individuals before initiation of hormone therapy. Identification of nonclassic CAH before initiation of hormone treatment in transgender is therefore important.
Little is known about the etiology of GD. It has been subject of increasing research within the fields of neuroanatomy, genetics, endocrinology, and psychiatry but a complete understanding has not been established so far. Twin studies suggest that genetic factors play a role (for a review 5 ) and prenatal exposure to T might also have an effect. 6
CAH is an autosomal recessive disorder with a reported incidence in the general population of 1:14.000-1:18.000. 4 A total of 95% to 99% of CAH cases are caused by mutations in CYP21A2 leading to varying degrees of 21-hydroxylase deficiency and consequently impaired cortisol and aldosterone biosynthesis and excessive androgen production. 7
In recent years, the association between GD and patients with known CAH has been studied,4,8,9 the hypotheses being that highly elevated androgens prenatally in female patients with CAH may influence gender identity. However, the number of studies is few, and some suggest an increased incidence of GD in patients with CAH while others do not (for review 6 ). Previous studies on the relationship between elevated androgens, as seen in CAH, and GD have not been congruent (for review 6 ). Some suggest that the prevalence of GD in patients with CAH appears to be higher than in the general population. Dessens et al. reported GD in 5.2% of patients with CAH reared as girls. 3 In comparison, the prevalence of GD in the general population in Denmark is estimated to be 0.05% for individuals who were assigned a male gender at birth and 0.05% for individuals who were assigned a female gender at birth in a recent study. 10 However, the prevalence of GD in the general population is uncertain and most likely underreported. To our knowledge, data on the prevalence of CAH among transgender individuals does not exist.
Conclusion
We here describe the coincident discovery of nonclassic CAH in 2 transgender individuals, 1 FtM and 1 MtF. We do not believe that the CAH in our patients was causally related to being transgender. These cases underline the importance of a thorough medical evaluation prior to initiating hormonal treatment as such finding may have influence on treatment options. In cases with suboptimal response to ACTH test, stress doses of hydrocortisone should be supplied during illness, physical stress, and surgery.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.