
Abstract

Transcriptome is the complete collection of RNA molecules produced by an organism’s genome under specific conditions or in a particular cell type. It includes all RNA types, such as mRNA, non-coding RNA (e.g., microRNA, long non-coding RNA), and various regulatory RNA. 1 The transcriptome provides a glimpse into gene expression within cells or tissues and can shed light on the dynamic regulatory processes that govern cellular functions. By analyzing RNA molecules within cells or organisms, researchers can identify active genes, measure their expression levels (gene expression profiling), and understand how various conditions or diseases impact gene expression. 1 This knowledge has been crucial in understanding cancer biology, such as identifying intrinsic subtypes of breast cancer and diffuse large B-cell lymphoma and providing consensus on molecular subtypes of colorectal cancer. Tools based on gene expression profiling, such as “Oncotype DX” and “Decipher,” provide insights into the risk of recurrence following breast and prostate cancer surgery, respectively. 2 Transcriptomics is a powerful tool crucial in detecting changes in coding sequences, including single nucleotide variants (SNVs), multiple nucleotide variants (MNVs), and structural variants. These changes can significantly affect gene product function and contribute to tumorigenesis, tumor progression, and therapeutic resistance in cancer biology. 2 By identifying these genomic alterations, transcriptomics can help uncover biomarkers for early cancer detection, prognosis prediction, and treatment response assessment. Furthermore, a comprehensive understanding of the transcriptomic landscape of cancer enables the development of innovative therapeutic approaches that target specific genetic aberrations, ultimately advancing cancer treatment strategies.
Amplification, a common occurrence in the human genome, often leads to increased oncogene expression, promoting tumor growth. Transcriptomics, combined with array-based comparative genomic hybridization, can efficiently detect copy number alterations and present a promising avenue for detecting copy number alterations as oncogenic drivers like HER2 amplification in breast and several other cancers. 3 This may help expand the role of antibody–drug conjugates, bivalents, or trivalents in cancer management.
While the role of translatable mRNA is well-established, a large portion of RNA does not translate into proteins but plays pivotal roles in human health and disease. Non-coding RNA, particularly long non-coding RNA (lncRNA), once considered transcriptional noise, has sparked significant interest in cancer research, offering a promising avenue for future breakthroughs. lncRNAs play many roles in cancer development and progression, serving as oncogenes or tumor suppressors and regulating various cellular processes associated with cancer pathogenesis. 4 Understanding the functions and mechanisms of action of lncRNAs can provide profound insights into cancer biology and potentially lead to the development of novel diagnostic and therapeutic strategies. Refer to Table 1 for some well-established lncRNAs involved in cancer development, showcasing cancer biology’s intricate and multifaceted nature. 5
LnCRNAs in Cancer.

Recent advancements in spatial and single-cell transcriptomics have transformed our understanding of cancer. Spatial transcriptomics integrates histopathological architecture with high-throughput sequencing, providing insights into tumors at a spatially resolved molecular level. 6 Single-cell transcriptomics allows for gene expression analysis at the individual cell level, revealing cellular heterogeneity and finding various cell types within tumors. Tumor heterogeneity can influence the aggressiveness of the cancer and its likelihood of spreading or recurring. 7 Understanding the extent and nature of heterogeneity within a tumor can help predict the prognosis for a patient, guiding decisions about treatment strategies and follow-up care. Heterogeneity plays a significant role in the development of drug resistance. While specific cancer cells may respond initially to treatment, others within the same tumor may be resistant, leading to treatment failure and disease progression. By characterizing tumor heterogeneity, researchers can identify potential mechanisms of drug resistance and develop strategies to overcome it, such as combination therapies or alternative treatment approaches.
MicroRNAs (miRNAs) are small, non-coding RNA molecules that play crucial roles in post-transcriptional gene regulation. While miRNAs usually regulate gene expression and maintain cellular homeostasis, dysregulation of miRNA expression or activity can contribute to oncogenesis.
8
Here is how miRNAs can become drivers of oncogenesis:
Overexpression of oncogenic miRNAs: Some miRNAs, known as oncomiRs, are overexpressed in cancer cells compared to normal cells. These oncomiRs can target and downregulate tumor suppressor genes, leading to uncontrolled cell proliferation, apoptosis inhibition, angiogenesis promotion, and metastasis enhancement. For example, miR-21 is often overexpressed in various cancers and promotes tumor growth and invasion by targeting multiple tumor suppressors. Under-expression of tumor suppressor miRNAs: Conversely, specific miRNAs act as tumor suppressors by inhibiting the expression of oncogenes or genes involved in promoting cancer progression. When the expression of these tumor suppressor miRNAs is reduced in cancer cells, the oncogenes they normally repress are no longer effectively regulated, leading to unchecked cellular proliferation and tumor growth. For instance, miR-34a is a tumor suppressor that targets cell cycle regulation, apoptosis, and metastasis genes. Under-expression of miR-34a is associated with poor prognosis in several cancer types. Dysregulation of signaling pathways: miRNAs can influence the activity of key signaling pathways involved in carcinogenesis, such as the PI3K/AKT/mTOR pathway, the Wnt/β-catenin pathway, and the TGF-β signaling pathway. Dysregulated miRNA expression can disrupt the balance of these pathways, promoting aberrant cell proliferation, survival, and metastasis. For example, miR-155 modulates the PI3K/AKT/mTOR pathway and promotes tumor growth and metastasis in various cancers. The dysregulation of miRNA is ascribed to the following: Epigenetic alterations, such as DNA methylation and histone modifications, can affect miRNAs’ expression in cancer cells. Aberrant DNA methylation patterns in the regulatory regions of miRNA genes can lead to the silencing of tumor suppressor miRNAs or the activation of oncomiRs, contributing to oncogenesis. Genomic alterations such as mutations, chromosomal amplifications, deletions, and translocations disrupt the regular expression or function of miRNAs in cancer cells. For example, amplifying the genomic locus encoding the miR-17-92 cluster has been observed in certain lymphomas, leading to overexpression of oncogenic miRNAs within the cluster and promoting tumor growth.
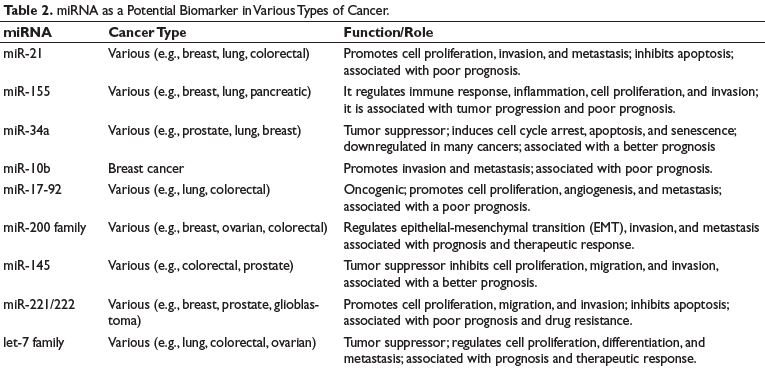
Overall, dysregulation of miRNA expression and activity can profoundly affect gene expression networks and signaling pathways implicated in cancer development and progression, making miRNAs important drivers of oncogenesis. Table 2 shows some examples of miRNAs, their physiological function, and their impact on cancer outcomes. 9
miRNA as a Potential Biomarker in Various Types of Cancer.
Circular RNAs (circRNAs) are a class of non-coding RNAs characterized by their circular structure, formed by back-splicing precursor mRNA transcripts. Emerging evidence suggests that circRNAs regulate gene expression by acting as microRNA sponges, sequestering miRNAs, and modulating their activity. Additionally, circRNAs regulate transcription by binding to regulatory proteins. 10 Dysregulated circRNA expression has been implicated in cancer development and progression, highlighting their potential as novel biomarkers for diagnosis, prognosis, and cancer therapy. 11 Table 3 shows some examples of circRNA and their role in various cancers.
CircRNA and Their Role in Different Types of Cancer.

Studying the roles of miRNAs and circRNAs in cancer transcriptomics offers further insights into the intricate regulatory networks underlying tumorigenesis. It provides new avenues for the development of targeted therapies and diagnostic strategies.
The field of transcriptomics employs advanced techniques such as RNA sequencing (RNA-seq) and microarray analysis to gain insights into gene expression. RNA-seq provides a comprehensive look at the transcriptome, allowing for precise measurements of gene expression levels and the discovery of new RNA species, including non-coding and alternative splicing variants.
On the other hand, microarray analysis offers a fast and efficient way to evaluate gene expression levels by hybridizing RNA samples on a chip with various probes that represent known genes. To access information about non-coding RNAs, researchers often turn to specialized bioinformatics tools and databases such as miRBase, circBase, and LNCipedia, which contain comprehensive records of annotated miRNAs, circRNAs, and lncRNAs, respectively. Integrative platforms such as TCGA (The Cancer Genome Atlas) and GTEx (Genotype-tissue Expression) provide transcriptomic data sets that cover coding and non-coding RNA expression profiles in various cancer types and normal tissues.
In conclusion, transcriptome analysis is a cornerstone in cancer management and therapy. Integrating transcriptomics with other omics data and clinical information holds immense promise for advancing precision medicine and patient outcomes. Ongoing validation studies aim to show the clinical utility of RNA-based non-invasive methods for cancer diagnosis, offering a hopeful glimpse into the future of cancer research and treatment.