
Abstract
Background
Previous evidence suggests serum concentrations of donepezil varies in clinical populations and that a dose higher than standard may have additional positive effect on cognition. Therapeutic drug monitoring (TDM) is a tool for dose optimization (DO) whereby treatment is adjusted based on previous quantification of the prescribed drug.
Objective
Investigate whether TDM-based DO of donepezil or memantine improves clinical outcomes and/or reduce the frequency of adverse reactions (ARs) in neurodegenerative conditions commonly treated with these two study drugs.
Methods
Single-blinded 1:1 randomized controlled study in an outpatient memory clinic. Eligible participants either newly diagnosed with Alzheimer's disease dementia (AD), dementia with Lewy bodies (DLB), or Parkinson's disease dementia (PDD) scheduled for treatment with donepezil or memantine. The intervention group received TDM based DO. The control group received DO solely based on clinical assessment. Clinical outcomes were change in Mini-Mental State Examination, Addenbrooke's Cognitive Examination, Neuropsychiatric Inventory, and Disability Assessment in Dementia from baseline to 12 months. Additionally, data on incidence and severity of ARs and proportion of participants with a serum concentration within the therapeutic reference range were collected.
Results
132 participants recruited (125 AD, 7 DLB, none with PDD) of whom 107 completed the study (101 AD and 6 DLB), fewer in the control group than planned. Statistical analysis did not reveal significant differences between groups neither for clinical outcomes nor for frequency of ARs.
Conclusions
TDM based DO did not significantly improve clinical outcomes nor reduce the frequency of ARs albeit important caveats to the results apply.
ClincialTrials.gov identifier
NCT04117178 (first posted October 7, 2019).
Keywords
Introduction
Therapeutic drug monitoring (TDM) refers to the procedure of quantification of serum or plasma concentrations of medications for subsequent dose optimization (DO). 1 Central to TDM is the concept of therapeutic reference range (TRR) within which the drug concentration is considered optimal. Below the TRR lower limit (LL) therapeutic response is unlikely, and at concentrations above the TRR upper limit (UL) tolerability decreases without further therapeutic benefits. 1 Recommendations for TDM in neuropsychiatry have been comprehensively reviewed by Heimke et al. 1
Since the approval and launch of donepezil, safety, tolerability and clinical efficacy of dosages of 15 mg, 20 mg, 2 and 23 mg daily (mg/d),3,4 have been compared to the standard treatment of 10 mg/d. A multi-center study with over 1300 patients with moderate-to-severe Alzheimer's disease dementia (AD) found a significantly improved cognitive benefit (using the Severe Impairment Battery (SIB) with a group difference of 2.2 points) for 23 mg/d dose compared to the standard dose of 10 mg/d. 3 In both studies investigating donepezil 23 mg/d versus 10 mg/d, the intervention group on donepezil 23 mg/d, had a significantly higher drop-out rate due to adverse reactions (ARs), compared to the standard dose of 10 mg/d (30.2% in 3 and 17.8% in 4 respectively).
A possible explanation for the findings of improved efficacy on SIB in AD patients on donepezil 23 mg/d compared to 10 mg/d3 is that a higher proportion of patients on the standard dose (10 mg/d) have subtherapeutic drug concentrations compared to patients on the high dose (23 mg/d). Also, the increased incidence of ARs in the group of patients treated with 23 mg/d compared to standard treatment could be explained by supratherapeutic concentrations of donepezil. Therefore, DO TDM could potentially improve clinical outcomes and reduce the frequency of ARs when drug dosing is tailored to the individual patient.
TDM for DO of anti-dementia drugs has only scarcely been investigated in a clinical setting. Hefner et al. 5 measured serum concentrations of donepezil in 106 patients diagnosed with AD. Results revealed highly variable serum concentrations among patients, even though all patients were treated with the standard dosage regimens, i.e., either 5 mg/d or 10 mg/d. Receiver operating characteristics (ROC) analysis based on the correlation between the overall clinical rating by the attending physician (Clinical Global Impression, CGI) and the drug concentration, revealed that the LL for the TRR was likely at least 50 ng/mL and thus above the previously stated LL at 30 ng/mL. 6 Ota et al. 7 reached a similar conclusion for the LL of the TRR for donepezil by in vivo inhibition of acetylcholine (AChE) in the brain using N-[11C] methylpiperidin-4-yl acetate ([11C]MP4A) positron emission tomography (PET).
The current guidelines for TDM recommends a TRR for memantine of 90–150 ng/mL 1 ; however, the evidence in support of this is sparse. No studies on TDM in treatment with memantine have correlated cognitive scores or data on global functioning to serum concentrations. Evidence rests primarily on single and multi-dose studies in healthy volunteers and participants with impaired renal function.8–10
In the present study we investigate the utility of TDM for donepezil and memantine in a randomized controlled design in a clinical setting. The study had two aims. The first was to determine whether individual DO of either donepezil or memantine based on TDM leads to a higher proportion of participants reaching a drug concentration within the TRR, compared to participants with no TDM guided dosing. The second aim was to investigate whether individual TDM guided DO improves cognition, activities of daily living (ADL), and neuropsychiatric symptoms compared to standard of care without TDM.
Methods
We conducted the study at the outpatient memory clinic at the Zealand University Hospital, Roskilde, Denmark. We screened patients with a clinical diagnosis of AD, dementia with Lewy bodies (DLB), or Parkinson's disease dementia (PDD) attending the memory clinic for study participation eligibility based on prior selected inclusion/exclusion criteria (Supplemental Table 1).
Inclusion and exclusion criteria
We chose the following inclusion criteria: the patient had to be recently diagnosed with either AD, DLB, or PDD and have a planned prescription of treatment with either donepezil or memantine. Among the exclusion criteria we chose were: patients not able or willing to give written informed consent (in the presence of a healthy relative), patients living alone not receiving help to administer medication, known psychiatric- or somatic condition possibly affecting cognitive performance, a history of drug or alcohol abuse, treatment with psychopharmacological agents possibly affecting cognition. For a full description of inclusion/exclusion criteria please visit Supplemental Table 1.
A senior neurologist (investigator PH) established the diagnosis of either AD, DLB, or PDD at a weekly consensus conference. AD was diagnosed according to the 2011 NIA-AA criteria, 11 DLB was diagnosed according to the 2017 DLB Consortium Criteria, 12 PDD according to the 2007 PD Movement Disorder Study task force report. 13 In the case that cognitive screening tests were not sufficient to reliably assess whether cognitive decline in a patient was severe enough to meet the criteria for dementia, a detailed neuropsychological examination was performed by a trained neuropsychologist. Thus, as per the 2011 NIA-AA criteria 11 a strict requirement for participation was significant impairment of at least two cognitive domains. In addition, symptom severity had to interfere with the ability to function at work or daily activities.
Participant recruitment and follow-up
An outline of the method and study visits is displayed in Figure 1.

Outline of the method and study visits. AD: Alzheimer's disease dementia; DLB: dementia with Lewy bodies: PDD, Parkinson's disease dementia: MMSE, Mini-Mental State Examination; ACE: Addenbrooke's Cognitive Examination; GDS: Geriatric Depression Scale; NPI: Neuropsychiatric Inventory; DAD: Disability Assessment in Dementia; S-donepezil: serum donepezil; S-memantine: serum memantine; cyp2D6: cytochrome P450 2D6 genotype; APOE: Apolipoprotein E; BchE-K: Butyrylcholinesterase K-variant; : blood sample collection;  : cognitive assessment.
: cognitive assessment.
Patients attending the memory clinic were offered study participation during the visit where the diagnosis and treatment options were presented. After a brief consideration period of 2–3 days, we invited the patient and relative to a (baseline) study visit during which the patient (from now on the participant) signed the informed consent and was then immediately randomized to either the control group (CG) or the intervention group (IG). The randomization procedure consisted in the participant drawing an envelope at random. Each envelope contained a note with a number from 1 to 150. The study nurse kept a randomization list specifying the group allocation of each number.
We conducted a baseline assessment of the cognitive impairment, depressive neuropsychiatric symptoms, and functional impairment. For the cognitive assessment, we used the validated Danish versions of the Mini-Mental State Examination (MMSE) 14 and Addenbrooke's Cognitive Examination (ACE) III. 15 For assessment of depressive symptoms, we administered the 15 item Geriatric Depression Scale (GDS) 16 to the participant. We conducted structured caregiver interviews using the Disability Assessment in Dementia (DAD) 17 and Neuropsychiatric Inventory (NPI). 18
At the 6-month visit, all participants had the regular assessments done by the neurologist according to the standard practice in the memory clinic. The regular assessment consisted of a clinical interview with the participant and primary relative focusing on symptoms of dementia and possible ARs to the study drugs along with the MMSE. In addition, all participants in the IG had a blood sample collected for analysis of the serum concentration of the treatment drug. All cognitive tests and caregiver interviews performed at baseline were repeated at the 12-month follow-up visit. In addition, we used the Clinical Global Impression Scale Improvement (CGI-I) and Clinical Global Impression Scale Severity (CGI-S) to assess global change of participant performance. 19
Reporting of adverse reactions
We used the EMA guidelines for good clinical practice E6 (R2) for reference on adverse events (AE) during the study. 20 We instructed participants and relatives to contact the clinic if an AR was suspected. A nurse specialist facilitated communication and scheduled visits for blood sample collection in case of a suspected AR. The primary Investigator (PI, investigator PH) oversaw assessment of both AEs and ARs and made all decisions on each participant whether dose optimization as per protocol was indicated (see Figure 2 for details). Participants who failed to complete the entire 12-month follow-up period on donepezil or memantine were registered as dropouts.
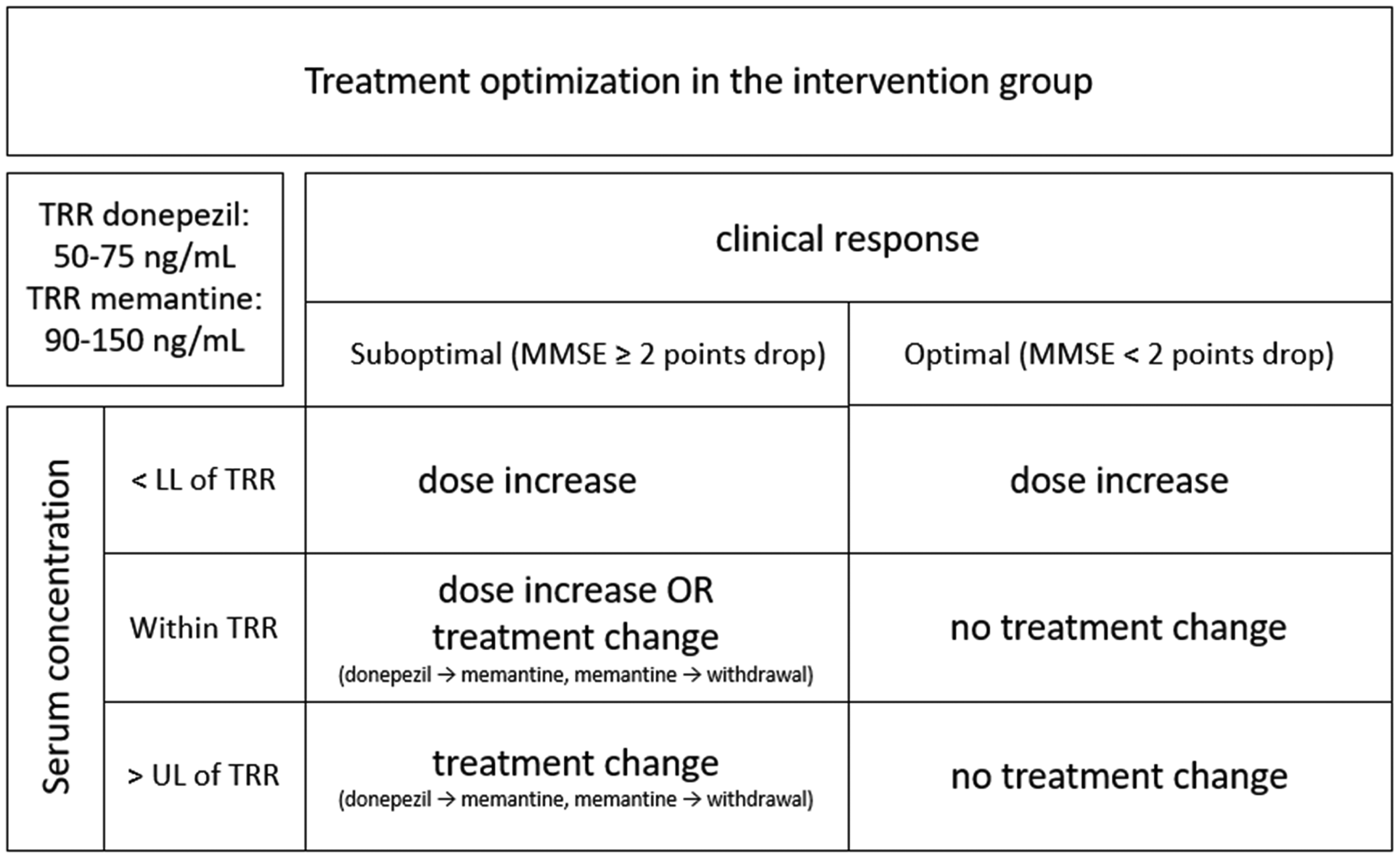
Treatment optimization in the intervention group.
Dose titration and optimization
At enrollment, all participants had their treatment titrated according to the standard of care at the study site. Hence, participants scheduled for donepezil were prescribed 5 mg/d for 4 weeks and then if well-tolerated the dose was increased to 10 mg/d. Participants scheduled for memantine were initially prescribed 5 mg/d, with a gradual dose-increase of 5 mg the following weeks to a maximum of 20 mg/d.
The PI assessed the effect of treatment on all participants at the regular 6-month visit and decided whether DO was indicated as per protocol. For the CG, treatment with the study drug was continued if well-tolerated and deemed reasonably efficacious based on the MMSE-score (a decline in score of <2 compared to baseline).
For the IG, the PI adjusted treatment at the 6-month visit according to the serum concentration of the study drug in addition to relevant clinical information identical to that of the CG. We presumed that the lower limit of the TRR for serum donepezil (S-donepezil) was 50 ng/mL as previously reported by Hefner et al. 5 Accordingly, for participants in the IG, if the 10 mg/d dosage was well-tolerated and the serum concentration was below the 50 ng/mL threshold we increased to daily dose to 15 or 20 mg/d. We applied the same procedure for treatment with memantine, with the lower limit set at serum memantine (S-memantine) 90 ng/mL. The maximum allowed daily dose in the study was 20 mg/d and 30 mg/d for donepezil and memantine respectively. In some instances, we did a second measurement of the serum concentration after the initial DO if the PI deemed it necessary.
We did serum concentration measurements for all participants in the IG whom we suspected of having an AR to determine whether an AR was due to a concentration of the study drug exceeding the upper limit of the TRR. 1 If an AR occurred, the PI adjusted the treatment to best allow for clinical efficacy and avoid future ARs. For the CG, this was done solely based on available clinical information, whereas for the IG the PI in addition relied on knowledge from prior serum concentration measurements.
We collected blood samples for serum concentration measurements from all participants at the 12-month visit. Based on the results hereof, the PI offered DO to participants in the CG according to the same principles as for the IG (see Figures 1 and 2). Also, if deemed to benefit the participants in the IG, DO was offered again.
One investigator (investigator MF) assessed the clinical outcomes of each participant. This investigator was kept blinded to the group allocation of the participant. In contrast, neither the PI who did DO during the study, nor the participants and their relatives were blinded to group allocation. Moreover, the statistical analyses were done in a completely blinded manner by an independent statistician (investigator VS) who was unaware of the group allocation of participants.
Laboratory methods and sample handling
We based the quantitative analysis of S-donepezil and S-memantine on the method published by Noetzli et al. 21 Blood samples were collected in fluoride citrate tubes. Quantification was done on a Waters Acquity UPLC connected to a Waters TQD mass spectrometer. Quantification relied on a single calibration point with regression through zero. Linearity (r2 > 0.99) was confirmed for donepezil when measured from 0 to 3500 nmol/L. We (investigators JBR and JBL) verified the precision of the analysis by exchanging samples, calibrators, and internal controls for parallel testing with the Laboratory of the Centre for Psychiatric Neuroscience, Department of Psychiatry, Lausanne University Hospital, Switzerland, headed by dr. Dr Chin B. Eap.
Outcome measures
TDM outcomes were: the proportion of participants, in the IG ad CG respectively, reaching a drug concentration within the TRR. Mean drug concentration of either donepezil or memantine at 12-month follow-up in the IG and GC.
Clinical outcomes were: comparison of IG and CG for change of score on MMSE, ACE, GDS, DAD, NPI, and CGI-I between baseline and 12-month follow-up. Change on ACE score from baseline to 12-month follow-up was the primary clinical outcome. In addition, we compared the CGI-S score between IG and CG at 12-month follow-up.
We compared the proportion of participants in the IG and CG experiencing common ARs. Furthermore, we investigated whether ARs in the IG were associated with a serum concentration above the UL of the TRR.
Statistics
Power calculation
Prior to study initiation we did a power calculation based on results from 5 and an unpublished pilot study conducted at the memory clinic. The results of these studies led us to presume that at least 50% of serum concentrations would be out of the recommended range (S-donepezil: 50–75 ng/mL, S-memantine: 90–150 ng/mL) without TDM based DO. Furthermore, we assumed that the use of TDM based DO would result in at least 75% of participants reaching a serum concentration within the TRR. We found that 55 participants were needed in each group to detect a group difference with a significance level of 5% and a power of 80% in a Z-test for absolute difference in proportions.
Statistical modelling for clinical outcomes
We assessed the TRR outcome at 12-month follow-up in a linear regression model with a binomial error term to better accommodate the binary outcome. Hence, the effect estimate is an absolute probability difference between randomization groups. The serum concentrations of either donepezil or memantine at 12-month follow-up were to be displayed in boxplots, but not evaluated in a statistical model as the two study drugs were not comparable in this setting.
We assessed the clinical outcomes at 12-month follow-up in linear regression models. Clinical outcomes were assessed using linear regression models comparing the difference between the baseline effect (difference in outcome between the randomization arms at study start) and the 12-month follow-up effect (difference in outcome between the randomization arms at the 12-month follow-up). We used generalized estimating equations (GEE) to accommodate for repeated measurements on the same study participant. We adjusted for possible bias due to potential differential dropout between groups by weighting the data points without missing values with the inverse probability of being in the data. 22 We estimated this probability from logistic regression models using baseline variables and assessments (sex, age, randomization group, MMSE, ACE, GDS, and NPI) as predictors.
Approvals
We conducted the study in accordance with the ethical standards of the 1964 Declaration of Helsinki and its later amendments. 23 Both the regional scientific ethics committee (Videnskabsetisk komité for Region Sjælland, file no.: SJ-596) and the Danish Medicines Agency approved the study (Lægemiddelstyrelsen, EudraCT-no.: 2017–002707-10) prior to initiation. The study protocol was approved by the local data supervisory authority (Datatilsynet Region Sjælland, approval no.: REG-007-2018). The Good Clinical Practice (GCP) unit of Copenhagen monitored the study externally and conducted regular site inspections. We registered and frequently updated information on the study on the website ‘clinicaltrials.gov’ (NCT = 04117178).
Results
Figure 3 provides an outline of screening and enrollment of participants.
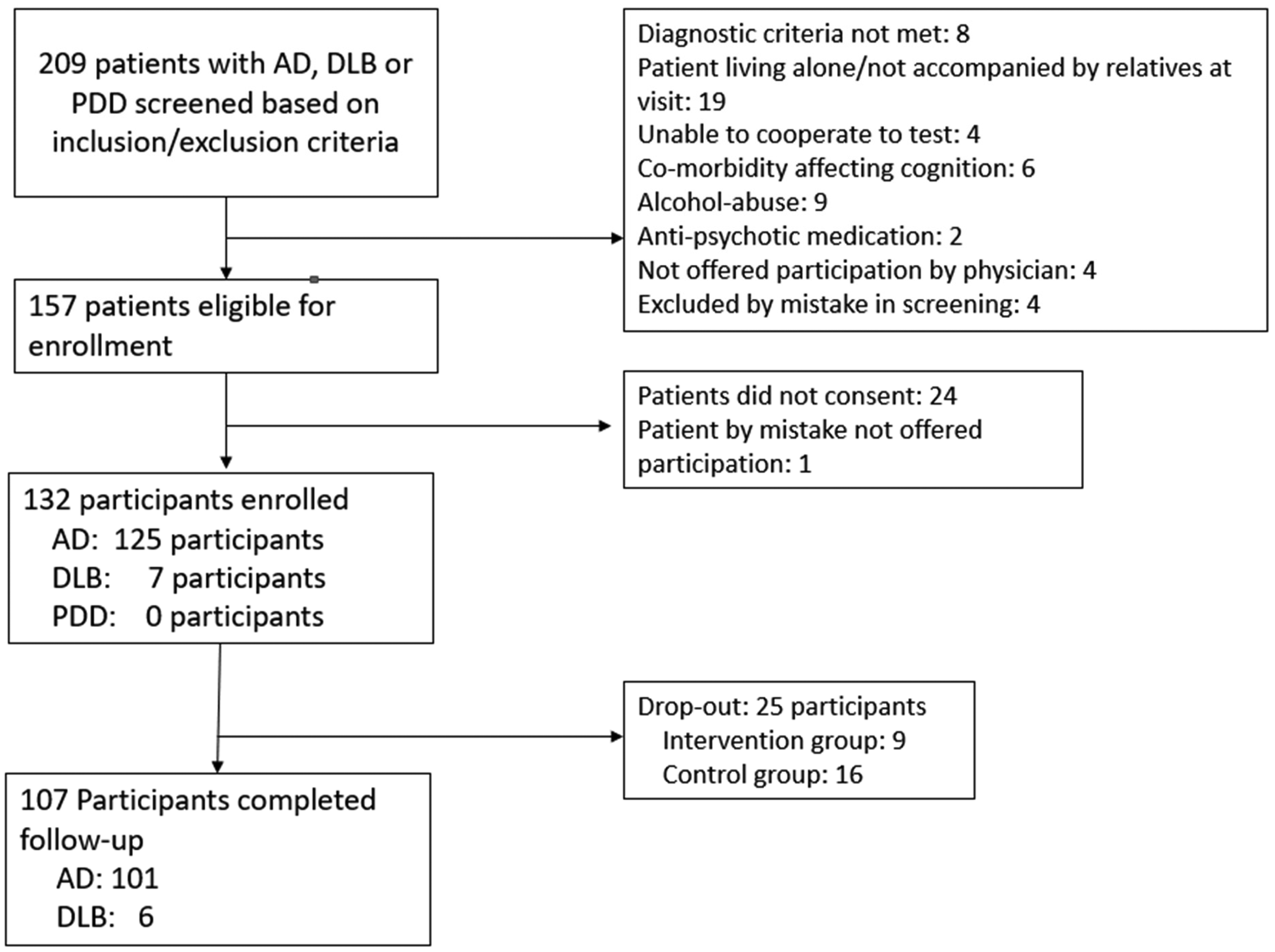
Outline of screening and enrollment of participants.
We enrolled a total of 132 participants from February 12, 2020, to February 24, 2022. Of the 132 participants, we registered 25 as drop-outs either due to a participant decision to withdraw or if change of treatment to an anti-dementia drug not investigated in this study (galantamine or rivastigmine). Of the 25 participants who withdrew from the study, 1 diagnosed with DLB and the rest diagnosed with AD. Although we did not find any significant group difference in the rate of dropout between IG and CG (Fisher's exact p = 0.12) we did note a slightly greater dropout rate in the CG compared to IG (16 of 65 versus 9 of 67 participants respectively). Importantly, dropout in the CG was higher than planned and resulted in a number of assessable participants at the 12-month visit (49) slightly below the projected number (55).
Details on withdrawal are summarized in Figure 4.
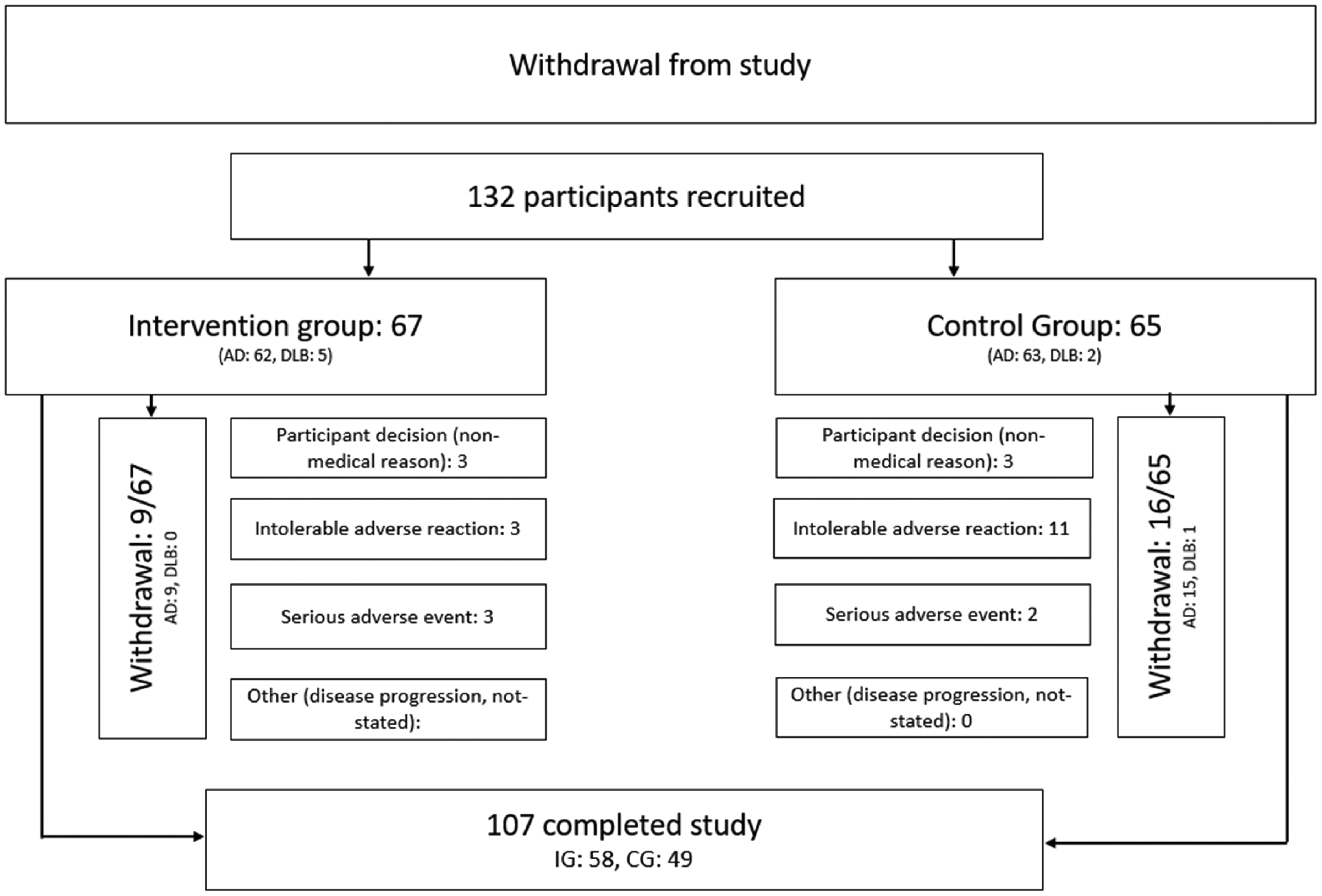
Details on participant withdrawal during the study.
Baseline characteristics of participants
At baseline, the IG consisted of 67 participants and the CG consisted of 65 participants. All demographic variables were equally distributed between the two groups except for sex. For this variable significantly more men were allocated to the IG relative to the CG.
Clinical outcomes
At baseline there was no statistical group difference on any of the clinical scores. Assessing the clinical outcomes (comparison of change of clinical scores between baseline and 12-month follow-up), we found no statistically significant differences between the two groups except for GDS. Here, we found a slight although significant difference (p < 0.01). The IG displayed a minor increase in GDS over the 12-month follow-up time whereas the CG displayed minor decrease in GDS.
Table 1 provides a summary of demographic variables and cognitive scores of enrolled participants at baseline.
Baseline characteristics of participants in the intervention and control groups.
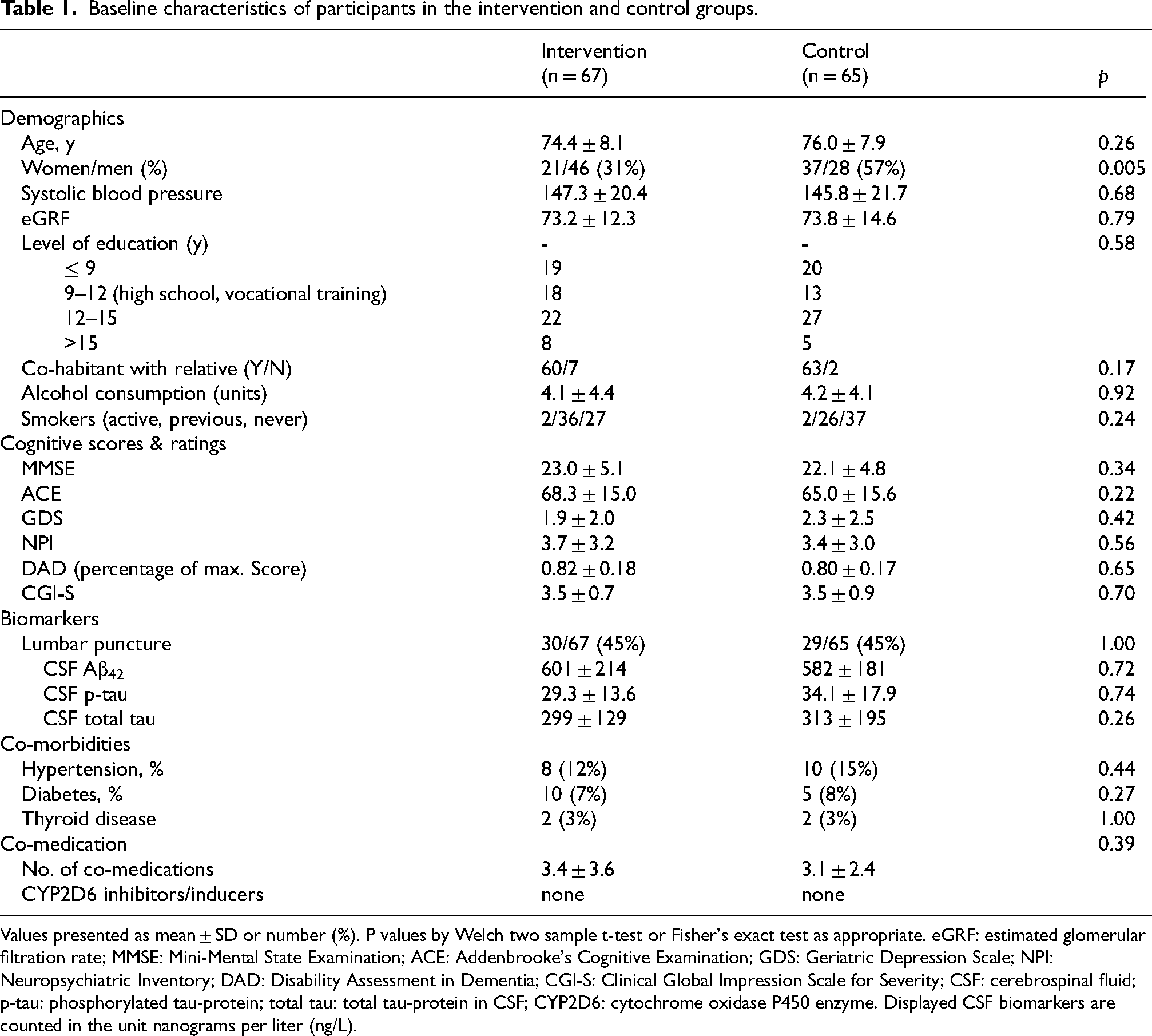
Values presented as mean ± SD or number (%). P values by Welch two sample t-test or Fisher's exact test as appropriate. eGRF: estimated glomerular filtration rate; MMSE: Mini-Mental State Examination; ACE: Addenbrooke's Cognitive Examination; GDS: Geriatric Depression Scale; NPI: Neuropsychiatric Inventory; DAD: Disability Assessment in Dementia; CGI-S: Clinical Global Impression Scale for Severity; CSF: cerebrospinal fluid; p-tau: phosphorylated tau-protein; total tau: total tau-protein in CSF; CYP2D6: cytochrome oxidase P450 enzyme. Displayed CSF biomarkers are counted in the unit nanograms per liter (ng/L).
Table 2 gives a summary of clinical outcomes for the IG and the CG respectively.
Changes in clinical outcomes from baseline to 1-year follow-up (N = 107).
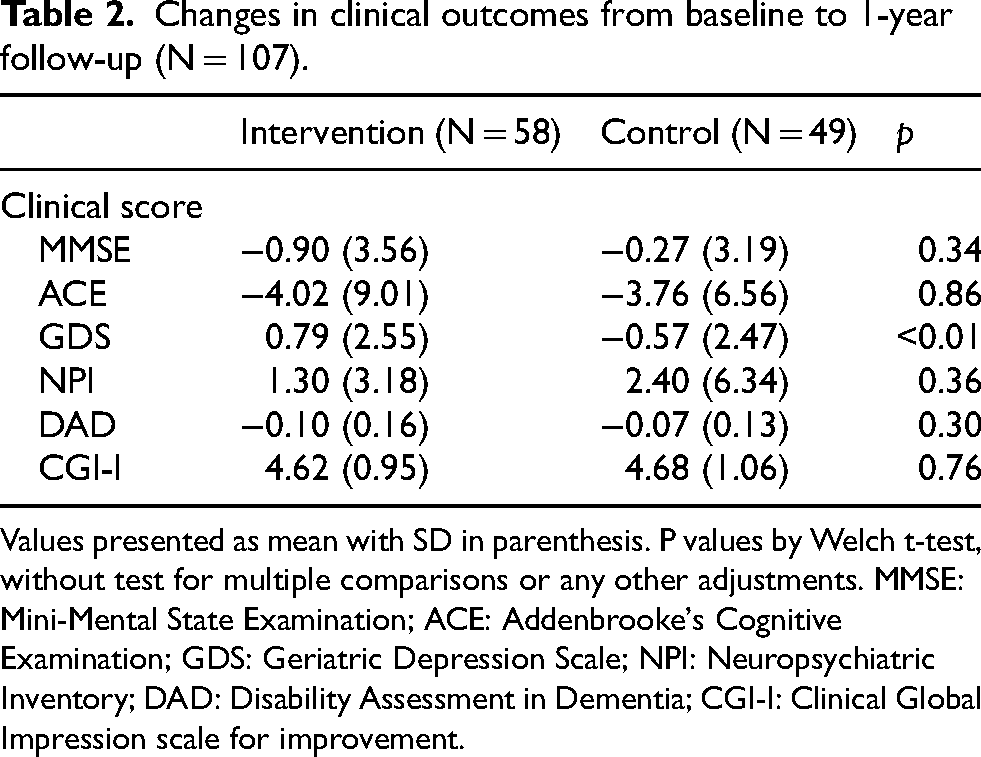
Values presented as mean with SD in parenthesis. P values by Welch t-test, without test for multiple comparisons or any other adjustments. MMSE: Mini-Mental State Examination; ACE: Addenbrooke's Cognitive Examination; GDS: Geriatric Depression Scale; NPI: Neuropsychiatric Inventory; DAD: Disability Assessment in Dementia; CGI-I: Clinical Global Impression scale for improvement.
Figure 5 is a visual display of clinical outcomes.
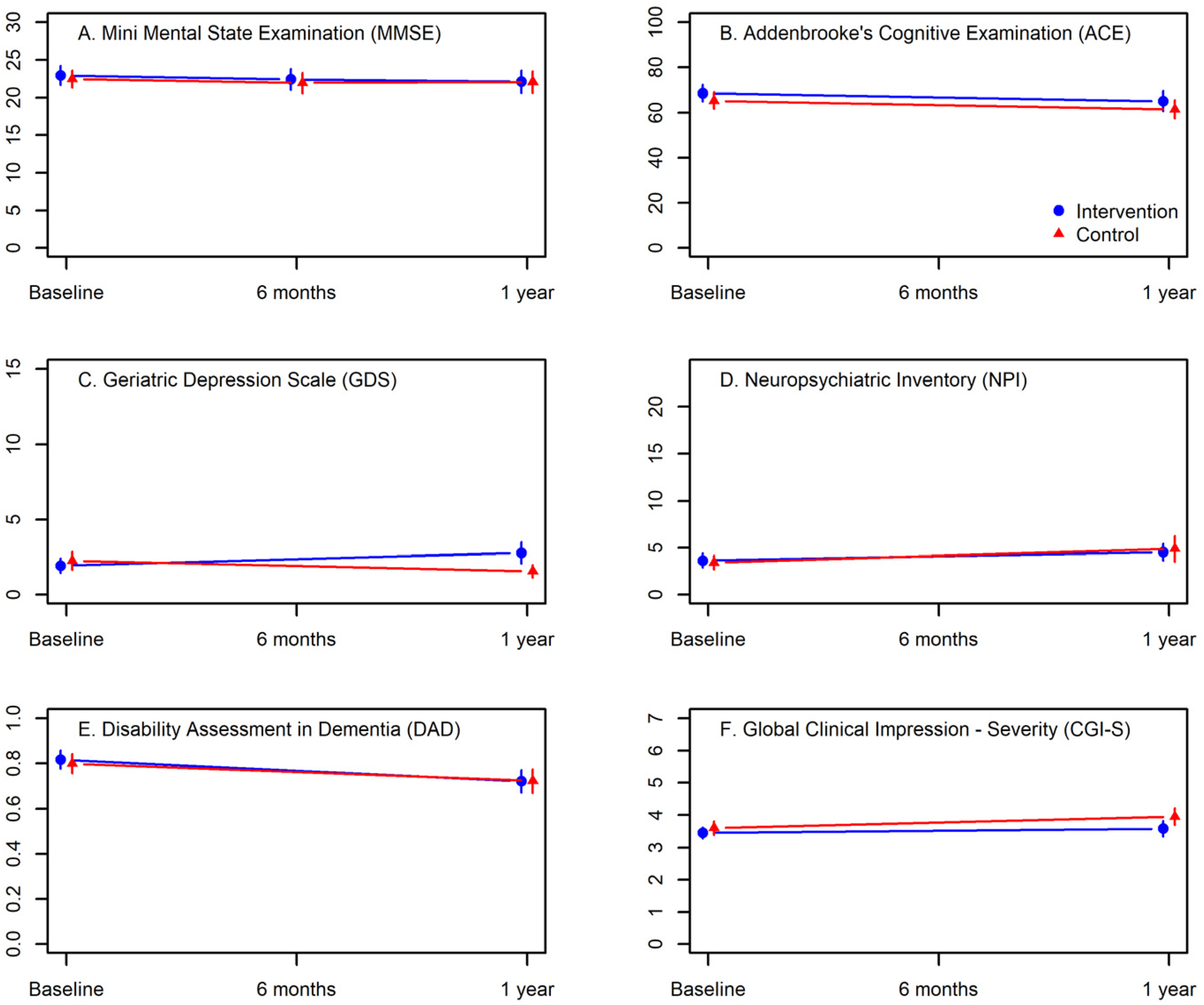
Visual display of clinical outcomes.
Optimized dose
The great majority of participants were initially prescribed donepezil. Only one participant in the IG and 1 participant in the CG were prescribed memantine from the outset. At the end of the study 99 participants received donepezil monotherapy, 5 participants received memantine monotherapy and 3 participants received a combination of the two study drugs. A total of 56 participants across IG and CG received DO during the study. In the IG, 38 of 67 participants had TDM based DO. Of the 38 in the IG who received TDM based DO 8 dropped out. In the CG, 18 participants had DO according to the standard of care treatment. Of these 18 participants in the CG 10 completed the study. After the 12-month visit, a total of 40 participants (39 participants on donepezil, 1 on memantine) received a daily dosage of donepezil or memantine above the standard dose (i.e., 10 mg or 20 mg once daily respectively).
Table 3 provides a summary on dose optimization in both the IG and the CG.
Summary on dose optimization in both the intervention and control groups.
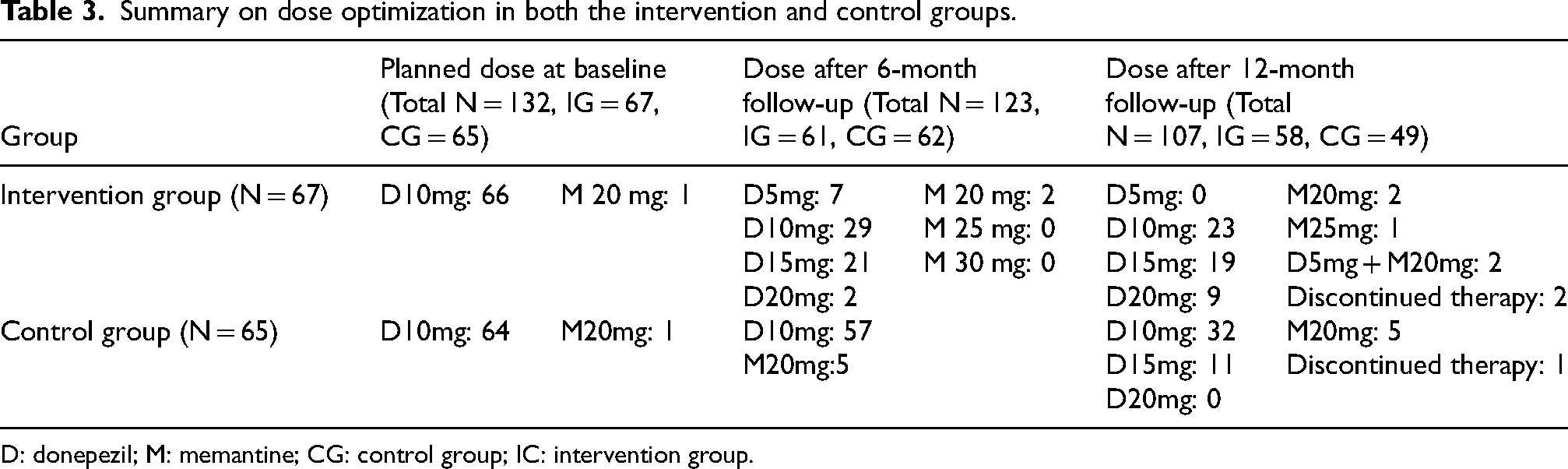
D: donepezil; M: memantine; CG: control group; IC: intervention group.
Adverse events and adverse reactions
During the study period, a total of 12 serious adverse events (SAEs) occurred in the IG and 10 SAEs occurred in the CG. One of the SAEs in the CG was an expected serious adverse reaction (SAR) due to accidental overdosing of donepezil on part of the participant. All other occurring SAEs were not related to the study drugs. Common non-serious ARs were reported for 41 participants, 22 in the IG and 19 in the CG. We found no significant difference in frequency of ARs between the two groups (Fisher's exact test p = 0.71). Of the 41 participants, 16 had a second AR and of these 5 had a third AR, of these 2 had a fourth AR. All ARs were mild to moderate in intensity and often self-limiting when treatment was continued. We registered dropout due to intolerable ARs for 3 participants in the IG and 11 in the CG (Fisher's exact test p = 0.02).
Table 4 provides a summary of ARs during the study in the intervention and control groups.
Common adverse reactions.
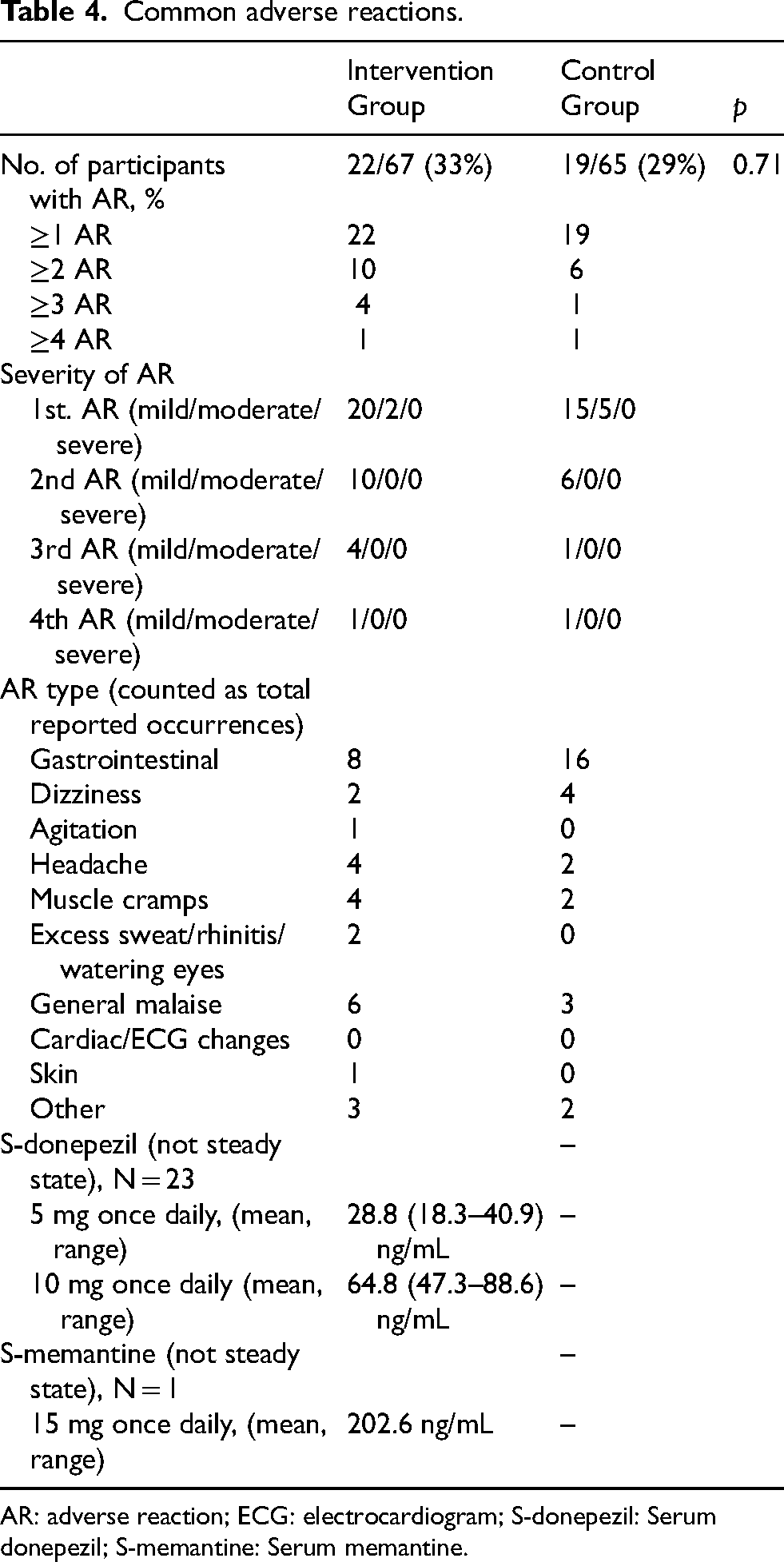
AR: adverse reaction; ECG: electrocardiogram; S-donepezil: Serum donepezil; S-memantine: Serum memantine.
TDM outcomes
At the 12-month follow-up visit, the proportion of participants with a serum concentration within the TRR was 39.9% in the IG and 48.0% in the CG. This 8.1 percentage point difference (CI95% [-11.6; 27.9]) was not statistically significant (p = 0.42).
At the 12-month visit, the IG had a mean S-donepezil of 58.4 ng/mL and a mean S-memantine of 245.7 ng/mL. The CG had a mean S-donepezil of 49.2 ng/mL and a mean S-memantine of 344.3 ng/mL.
Figure 6 shows the distribution of serum concentrations for donepezil and memantine at the 12-month follow-up visit.
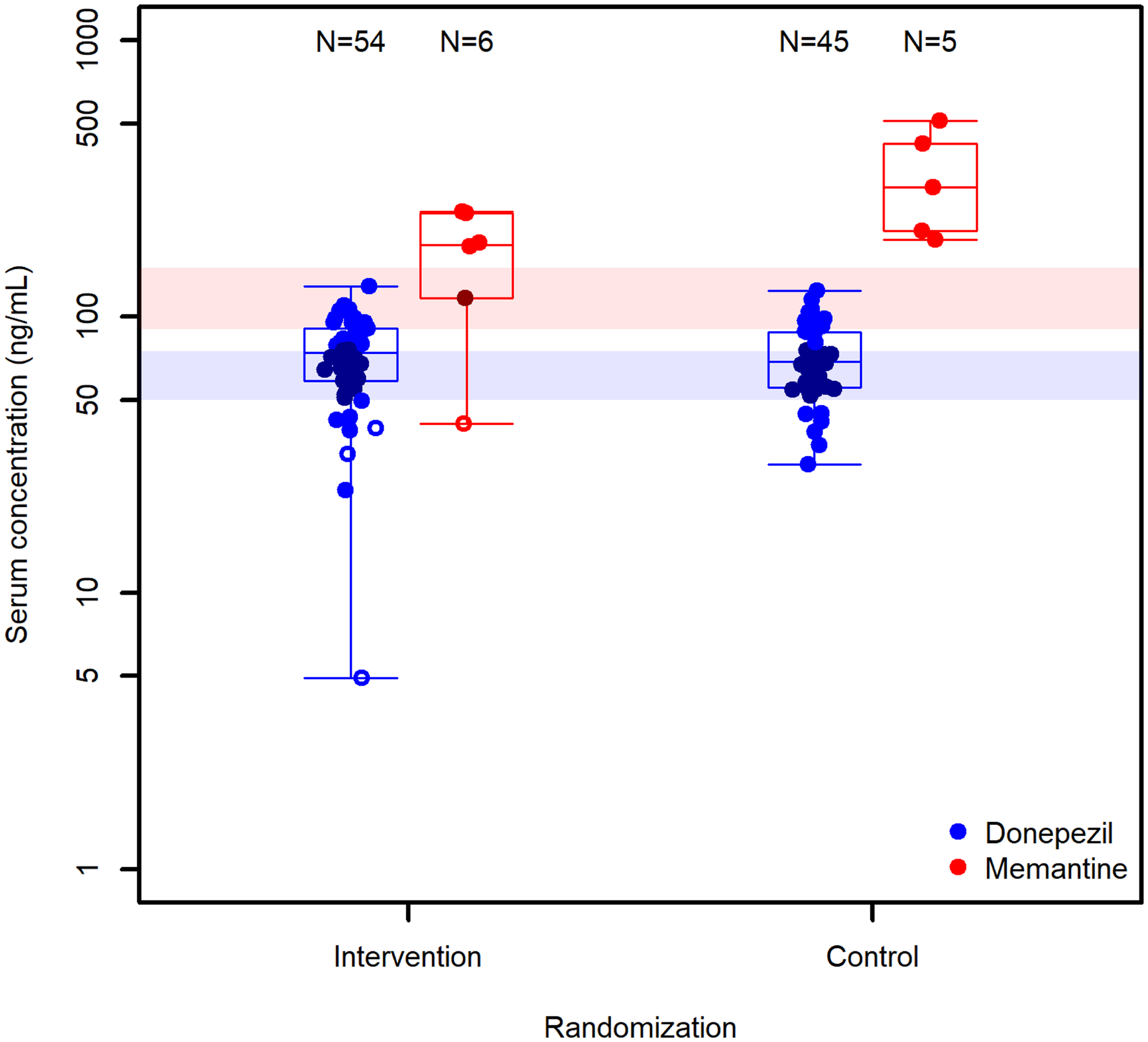
Boxplots showing the distribution of serum concentrations for donepezil and memantine at the 12-month follow-up visit. x-axis: group allocation of participants, either intervention group (left) or control group (right); y-axis: measured serum concentration of study drug in nanogram per milliliter; Blue color dots: serum donepezil for participants adherent to therapy; Blue color circles: serum donepezil for participants poorly adherent to therapy; Red dots: serum memantine for participants adherent to therapy; Opaque blue colored bar: therapeutic reference range for donepezil; Opaque red colored bar: therapeutic reference range for memantine.
Discussion
The present study is the first randomized controlled study to investigate the utility of TDM for DO of anti-dementia drugs in a clinical setting. Of the enrolled 132 participants 56 had received DO based on TDM during or after the conclusion of the study. However, we were not able to detect a significant group difference for neither cognition, ADL-functioning, neuropsychiatric symptoms nor global functioning between the IG receiving DO of therapy based on TDM compared to the CG receiving standard care.
The results of the present study could not confirm the results from previous studies on TDM in AD. Importantly though, the small sample size of patients on memantine in this study warrants caution in drawing conclusions about TDM in this subgroup.
Hefner et al. 5 reported a significant difference of the likelihood of a good clinical outcome assessed by the CGI between participants having a S-donepezil below or above 50 ng/mL. Hefner et al. 5 performed a ROC analysis to determine the best cut-off for serum donepezil between a favorable and a less favorable outcome on the CGI, but data for this analysis data was only available for 38 participants.
In a prospective naturalistic setting with 42 participants, most diagnosed with AD, Miranda et al. reported a slightly higher mean S-donepezil for ‘good’ versus ‘bad’ responders to donepezil therapy using the MMSE. 24 Both the study by Hefner et al. 5 and the study by Miranda et al. 24 had an observational design, simple outcome measures and relatively small sample sizes, thus positive results due to chance are plausible.
Contrary, the significant results on cognitive functioning (on SIB) reported by Farlow et al. 3 comparing donepezil 23 mg/d to 10 mg/d was based on a randomized controlled trial with a study population of more than 1300 patients. Here, a post-hoc analysis suggested that the increased cognitive effect of the 23 mg/d dose was more evident in patients with more advanced disease. Although, the results on cognition could not be reproduced in a smaller subsequent study, 4 the patient population in both studies consisted in patients with more severe AD (eligible MMSE score was 0–20 and 0–12 respectively) and they are therefore not directly comparable to the present study.
In the present study, we found an overall frequency of ARs in both the IG and CG about 30%. The overall withdrawal rate due to ARs was 11% which is similar to previous studies standard doses of donepezil.25,26 Of note, a significantly higher number of participants in the CG withdrew from the study due to intolerable ARs compared to the IG (p = 0.02). However, the absolute numbers were very small (3 out of 67 versus 11 out of 65 participants. Furthermore, participants themselves were not blinded to group allocation which may suggest a higher willingness to complete the study for participants in the IG despite experiencing ARs. We collected a total of 24 blood samples from participants who had experienced common ARs to investigate whether ARs were caused by abnormally high serum concentrations (i.e., above the UL of the TRR). In only one sample, collected from a participant treated with donepezil 10 mg daily, we found a S-donepezil slightly above the UL of the TRR. Thus, common ARs on standard doses of donepezil are likely not due to the serum concentration being above the UL of the TRR.
A meta-analysis of a total of 30 clinical studies on donepezil in AD found a higher frequency AEs including ARs, in patients treated with donepezil 5 mg/d or 10 mg/d compared to placebo. 27 Also, the rates of nausea, diarrhea, and vomiting were significantly higher in patients treated with 10 mg compared to 5 mg. 27 Both abovementioned studies comparing donepezil 23 mg/d to 10 mg/d reported a higher frequency of ARs in the 23 mg dose group compared to the 10 mg dose group.3,4 Hence, the general risk of ARs is related to S-donepezil but on an individual level, results from the present study do not support a role of TDM in prediction and management of ARs. Factors influencing the risk of ARs could be inter-patient variability in the sensitivity to alterations of cholinergic signaling both in peripheral tissues 28 and the central nervous system making some patients prone to ARs at low serum concentrations while others tolerate donepezil at serum concentrations well above the TRR. Potential future research is the study of individual differences in cholinergic signaling and tolerability of acetylcholinesterase inhibitors.
We collected only one blood sample from a participant experiencing an AR on memantine. Consequently, we cannot reliably assess whether ARs on memantine are caused by serum concentrations being above the UL of the TRR. Nevertheless, in 12 out of 13 measurements of S-memantine at routine study visits, the concentration was above the UL of the TRR. Consisting with previous results, 29 many patients seem to tolerate the drug well above the UL of the TRR.
This study had several limitations which may explain the non-significant results of using TDM guided DO on clinical outcomes.
As evident from Table 1, the randomization procedure did not result in an equal sex distribution among groups, with the IG having a significantly higher proportion of men compared to the CG and vice versa. This must be the result of chance because the randomization procedure was correctly executed and strictly adhered and reviewed by external monitors. The unadjusted statistical analysis did not reveal significant differences for clinical outcomes between groups. To strengthen this finding, we did a post hoc baseline adjustment to mitigate potential confounding due to differences in sex distribution with the same results.
It can reasonably be assumed participants with more severe cognitive decline are also more likely to drop out from a study before cognitive assessment and intervention benefit for dropouts will not be registered. Therefore, a direct comparison between groups would likely result in a conservative estimate of intervention effect. In this study, we sought to reduce the effect of differential drop out by adjusting the statistical analysis for factors likely to impact group differences in dropout rates. We chose a number of known factors likely to explain differences in dropout, however, other factors impacting dropout may still be present and thus left un-accounted for.
A slight difference in change on the GDS was noted between the IG and the CG. Albeit adjustment for difference in GDS between groups, depressive symptoms are known to impact cognition and could therefore conceal an effect. The observed significant difference in GDS at 12-month follow-up can be explained by a slight group difference in cognitive score as depressive symptoms are known to decrease as AD progresses.30,31 Although the rate of cognitive decline in the two groups was of a similar magnitude, both at baseline and at 12-month follow-up the IG had a slightly higher mean MMSE and ACE.
On the assumption that participants with a subtherapeutic serum concentration (i.e., below the LL of the TRR) had some drug effect, the effect size of TDM based adjustment could be too small to allow for statistically significant effect of the intervention. It must also be stressed, that for a large part of participants in the IG (42%) TDM did not justify DO.
Most participants in the IG had TDM done only once during the study at the 6-month visit. The result of TDM (i.e., the measured serum concentration) is likely influenced by several factors, some of which may display intra-participant variability (time from ingestion, co-medication, weight, treatment adherence, etc.). Although, we did our best to take such information into account when performing DO, ideally TDM should be performed more than once per participant to ensure that the dose is indeed optimal.
A concern with TDM in general is that TDM does not by itself inform how to dose optimally. 32 Commonly, the TRR is defined as the arithmetic mean ± standard deviation of the concentrations in blood of ‘responders’ when evidence in support of the TRR limits is sparse. 1 Still, this still leaves out the question of how-to assess treatment response in dementia. Clinical outcomes can be influenced by many other factors apart from the serum concentration of the treatment drug. Therefore, ideally drug response should be assessed objectively by reliable biomarkers in addition to clinical outcomes. The quantitative electroencephalography feature alpha band magnitude-squared coherence (MSCOH) has been shown to be decreased in AD compared to heathy controls 33 and since anticholinergic drugs have been shown to decrease MSCOH in heathy controls, 34 MSCOH could be a possible biomarker for the cholinergic effect of donepezil, but whether there is a dose-response relationship of donepezil upon MSCOH in AD has yet to be investigated.
In conclusion, this study did not support general usage of TDM based DO for donepezil and memantine in dementia, albeit important study limitations and caveats to the external validity of the results apply.
Supplemental Material
sj-docx-1-alr-10.1177_25424823241289373 - Supplemental material for Therapeutic drug monitoring for dose optimization in Alzheimer's disease and in dementia with Lewy bodies: A randomized single-blinded clinical trial
Supplemental material, sj-docx-1-alr-10.1177_25424823241289373 for Therapeutic drug monitoring for dose optimization in Alzheimer's disease and in dementia with Lewy bodies: A randomized single-blinded clinical trial by Michael Hén Forbord Fischer, Ivan Chrilles Zibrandtsen, Peter Johannsen, Volkert Siersma, Jan Borg Rasmussen, Jens Borggaard Larsen and Peter Høgh in Journal of Alzheimer's Disease Reports
Footnotes
Acknowledgments
We would like to thank specialist nurse Susanne Kristiansen, laboratory technicians Jane Hansen Damm and Anja Rikke Sjelhøj for their invaluable help in planning and executing this study.
Author contributions
Michael Hén Forbord Fischer (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Visualization; Writing – original draft; Writing – review & editing); Ivan Chrilles Zibrandtsen (Conceptualization; Investigation; Methodology; Supervision; Writing – original draft; Writing – review & editing); Peter Johannsen (Conceptualization; Formal analysis; Methodology; Supervision; Writing – original draft; Writing – review & editing); Volkert Siersma (Formal analysis; Investigation; Methodology; Software; Visualization; Writing – original draft; Writing – review & editing); Jan Borg Rasmussen (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Validation; Writing – original draft; Writing – review & editing); Jens Borggaard Larsen (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Validation; Writing – original draft; Writing – review & editing); Peter Høgh (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research project was supported by the Danish Alzheimer's Disease Research Foundation (grant no. 670-181003) and the Department for Clinical Medicine, University of Copenhagen.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available (in an anonymous version) on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.