
Abstract
Bestrophinopathies are a group of clinically distinct inherited retinal dystrophies that typically affect the macular region, an area synonymous with central high acuity vision. This spectrum of disorders is caused by mutations in bestrophin1 (BEST1), a protein thought to act as a Ca2+-activated Cl- channel in the retinal pigment epithelium (RPE) of the eye. Although bestrophinopathies are rare, over 250 individual pathological mutations have been identified in the BEST1 gene, with many reported to have various clinical expressivity and incomplete penetrance. With no current clinical treatments available for patients with bestrophinopathies, understanding the role of BEST1 in cells and the pathological pathways underlying disease has become a priority. Induced pluripotent stem cell (iPSC) technology is helping to uncover disease mechanisms and develop treatments for RPE diseases, like bestrophinopathies. Here, we provide a comprehensive review of the pathophysiology of bestrophinopathies and highlight how patient-derived iPSC-RPE are being used to test new genomic therapies in vitro.
Introduction
Pathogenic mutations in BEST1 are known to cause a number of distinct autosomal dominant dystrophies including Best disease, Adult vitelliform macular degeneration (AVMD), autosomal dominant vitreoretinochoroidopathy (ADVIRC) and the recessive disease, autosomal recessive bestrophinopathy (ARB).1,2 This spectrum of diseases, collectively known as bestrophinopathies, affect the macular region, with patients typically presenting in clinic within the first two decades of life. The prevalence of individual bestrophinopathies is rare and varied, however, in a recent study of 3000 patient families with inherited disease, BEST1 was the 5th most mutated gene, accounting for 3.9% of affected families, and one of the highest causes of autosomal dominant macular dystrophy. 3 Bestrophinopathies primarily affect the retinal pigment epithelium (RPE), a monolayer of cells that interacts with and sustains the light responsive retina. In the RPE, BEST1 is normally expressed on the basolateral membrane, where it acts as an ion channel.4–6 Dysfunction of RPE with age or as a result of inherited mutations, can lead to degeneration of the retina. Over the last 30 years, research has focused on investigating the biological function of BEST1 in the RPE and the mechanisms by which BEST1 mutations contribute to retinal disease. These advances have led to a better understanding of disease mechanisms and development of potential therapies to treat the unmet clinical needs for bestrophinopathies.
Clinical spectrum of bestrophinopathies
Best disease
Best disease is the most common bestrophinopathy, with an estimated prevalence ranging from 1:5,000 to 1:67,000 and an onset that usually occurs during childhood or early adulthood.7,8 The disease was first described in 1883 as a peculiar change in the macula 9 and later identified in a large German family, by Dr Friedrich Best, who noted its dominant inheritance pattern. 10 Best disease is characterised by the presence of vitelliform lesions that evolve over time, and progress through a number of stages, which may or may not follow chronologically for each patient. During the initial previtelliform stage, the fundus appearance and visual acuity (VA) are normal; however, there may be subtle changes of the RPE, appearing on the central retina as yellowish pigment changes, with some granularity defects.2,11 Best disease is most easily identified during the vitelliform stage, where a yellow demarcated ‘egg-yolk’ vitelliform lesion, 2–3 mm in diameter, is observed on the macula (Figure 1(a)). At the vitelliform stage there may be mild vision loss and a decrease in VA, although VA has been reported to be normal in some patients.2,11 Patients may also report photophobia, metamorphosis and night blindness at this stage.7,12

Representative illustrations of Bestrophinopathy fundus appearance. (a) Best disease, with the egg yolk-like vitelliform lesion observed at the macula. (b) Autosomal recessive bestrophinopathy, characterised by multifocal deposits and lesions around and beyond the macula. (c) Autosomal dominant vitreoretinochoroidopathy typified by presence of a hyperpigmented circumferential band of pigmentation in the peripheral retina. (d) Best-related retinitis pigmentosa characterised by the presence of peripheral pigment changes, bone spicules and foveal deposits.
Over time, the demarcated borders and yellow appearance of the vitelliform lesion can become more irregular and a partial resorption of fluid may lead to the appearance of a pseudohypopyon, where various inflammatory cells infiltrate the anterior chamber. Despite the dramatic appearance of a pseudohypopyon, VA may only be mildly affected.2,11 The disease can then progress to a vitelliruptive stage, where breakdown of the vitelliform lesion leads to a ‘scrambled-egg’ fundus appearance, with irregular yellow deposits present. At this stage, VA may be steady or may begin to decline as the disease progresses.2,13 Best disease can then progress to an atrophic stage, resulting in death of the RPE and loss of photoreceptors cells. Hyperpigmented fibrous scar tissue can be present in the macula, leading to widespread geographic atrophy, synonymous with progressive and irreversible retinal cell loss. 11
During the atrophic stage VA can decline dramatically (from 6/12 to 6/60 or less) and loss is irreversible. The breakdown of the RPE barrier can also lead to choroid neovascularisation (CNV). The presence of newly created, weak blood vessels, prone to rupturing, can lead to subretinal bleeds, fluid accumulation and a sudden decrease in VA. 14 Complications due to CNV have been reported to occur in 2%–9% of Best disease patients. 7 Best disease is generally bilateral, with lesions displaying some relative symmetry, but cases of unilateral Best disease have also been described. 15
For the most part, vision loss is gradual with 75% of affected individuals retaining a VA of 6/12 or better into their fifties in at least one eye; 13 however, the presence of CNV can lead to a steep decline in VA. Patients can also present with hyperopia, astigmatism 16 and anterior segment abnormalities, such as shallow anterior chambers, putting them in the risk group for having a narrow anterior angle and suffering from acute angle closure glaucoma (AACG). Best disease can also be multifocal, with numerous vitelliform lesions varying in size clustered around the macular. 17 Patients can also be prone to subretinal haemorrhages, the formation of macular holes, and retinal detachment in response to modest trauma.7,18
Adult vitelliform macular degeneration
Adult vitelliform macular degeneration is a dominantly inherited bestrophinopathy. AVMD typically has an onset of between 30 and 50 years of age, with more females seeming to be affected. 12 AVMD patients present with vitelliform lesions (similar to Figure 1(a)) that can be smaller than those observed in Best disease patients. Generally, AVMD has a less dramatic effect on vision and patients may not progress through all the stages described for Best disease, however, although CNV is rare, atrophy can occur 7 with complications, such as CNV or pigment epithelial detachment.19,20 In general, AVMD is clinically indistinguishable from Best disease and has been described as a milder form of Best disease, owing to its later onset and slower progression. The resemblance between the two conditions has led to the suggestion that AVMD should be reclassified as Best disease.2,7
Autosomal recessive bestrophinopathy
ARB is an autosomal recessive disease resulting from bi-allelic homozygous or compound heterozygous mutations within the BEST1 locus. Parents of ARB patients generally have normal vision, suggesting a tolerance of BEST1 haploinsufficiency.7,21 ARB is estimated to have a prevalence of 1:1,000,000, with an onset range between 4 and 40 years of age, although a juvenile onset is typical.7,22 ARB presents distinctively, with an alteration in the RPE leading to the formation of multiple subretinal deposit at the macula and midperipheral retina 21 (Figure 1(b)). The distinctive vitelliform macular lesions seen in Best disease are rare but may be present in some ARB patients at late stage. 23 The fundus of patients with ARB has a speckled appearance with multiple yellow/white, round, demarcated, and partially confluent lesions located towards the fovea and the vascular arcades, at the posterior pole and around the optic nerve.21,24 ARB also appears to affect the periphery, indicated by presence of peripheral drusen and RPE atrophy. 23
Patients also typically present with accumulation of subretinal fluid, culminating in some patients having cystoid macular edema with retinal fibrosis.7,22,25,26 Some developmental anomalies may also be present in ARB, and patients may be hyperopic and have shallow anterior chambers, increasing their risk of AACG.7,21 Vision loss is slow and progressive, 23 but in some cases central vision may eventually stabilise; however, complications such as CNV and AACG can lead to a rapid deterioration of vision.7,21,24,27
Autosomal dominant vitreoretinochoroidopathy
ADVIRC follows a dominant mode of inheritance and seems to differ from the other bestrophinopathies as it does not present with distinctive lesions of the macula. Instead, ADVIRC patients present with a strongly demarcated 360-degree circumferential hyperpigmented band in the peripheral retina, from the equator to the ora serrata28,29 (Figure 1(c)). ADVIRC is a very rare condition with a prevalence of 1;1,000,000, with only five BEST1 mutations currently known to cause the disease.28,30 The typical age of onset is during early childhood, with the hyperpigmented band generally seen in early stage patients. For older patients, the hyperpigmented band is considered a hallmark for ADVIRC; however, a recent reports have described its absence in some patients, indicating a high phenotypic variability.2,31,32
During early stage disease, ADVIRC affects the peripheral retina, with very little change in the appearance of the central retina. As ADVIRC progresses, it can encroach onto the macular region, causing central vision loss. 33 VA can range from 6/6, to absence of light perception in some cases; however, the majority of patients are able to maintain good vision of at least 6/12 throughout life.31,33,34 Patients can also present with punctate white retinal opacities, a pale optic disc and attenuated and narrow blood vessels.33,34 Further complications include retinal neovascularisation (leading to retinal bleeding and macula edema), retinal fibrosis, atrophy of the underlying choroid, retinal detachment and vitreous haemorrhage.7,28,33,35 Interestingly, ADVIRC patients may also have a wide range of eye development issues, including: microcornea, nanopthalmos, discrete rotatory nystagmus, hyperopia, presenile cataracts, iris dysgenesis, optic nerve dysplasia and a shallow anterior chamber leading to risk of AACG.7,28,33–35 These findings suggest that bestrophinopathy mutations may also contribute to broader ocular defects. Microcornea, rod-cone dystrophy and staphyloma (MRCS), a disease linked to BEST1 mutations, shares many ADVIRC-associated clinical features and the phenotype could be on the spectrum of ADVIRC disease expressivity.28,36
Retinitis pigmentosa
A number of patients have also been diagnosed with a bestrophinopathy-related form of retinitis pigmentosa, 37 classified by dense pigmentary changes in the peripheral retina. This can be accompanied by retinal gliosis, vascular attenuation, peripheral bone spicules, pale optic discs, yellow and foveal deposits and macular edema (Figure 1(d)), leading to patients typically having vision loss and night blindness. However, these may represent misdiagnosed cases of ADVIRC or ARB.1,2,38 Targeted next-generation sequencing has retrospectively diagnosed cases of bestrophinopathy-related RP as ADVIRC, despite patients lacking the hallmark ADVIRC hyperpigmented circumferential band on clinical presentation. 32 Alternatively, BEST1-related RP may be the result of multigenic mutations.39,40
Diagnosis of bestrophinopathies
Although the clinically distinct features of the bestrophinopathies may be sufficient for diagnosis, a number of additional tests can be used for confirmation. The electrooculogram (EOG) measures the standing potential, the electrical difference between the front and the back of the eye, and is used to assess the potential across the RPE, an indicator of its health. 41 The EOG measures the potential during exposure to the dark and following exposure to light, to calculate a light peak (LP): dark trough ratio, termed the Arden ratio. 41 According to the International Society for Clinical Electrophysiology of Vision (https://iscev.wildapricot.org/standards), a standard Arden ratio is between 1.7 and 4.3; this typically decreases below 1.5 in patients with bestrophinopathies.21,23,28,33,35,41 However, in some patients, the Arden ratio may be normal or only slightly reduced,11,12,33,42 this has been observed in 8% of Best disease cases, 31 highlighting the need for genetic testing to correctly distinguish and diagnose a bestrophinopathy. The electroretinogram (ERG) is typically found to be normal in patients, although delayed cone and rod response may be observed in ADVIRC patients28,31,34 and full-field, pattern and multifocal ERG’s can be affected in ARB patients.2,24,27,43
Disease progression can be monitored using optical coherence tomography (OCT), measuring foveal thickness, retinal degeneration and RPE atrophy, and the presence of retinal edema or subretinal fluid.7,16,23,35 Fluorescein angiography and fundus autofluorescence, can also be used to examine blood vessel structure and accumulation of lipofuscin respectively in patients. 7 Hyperfluorescence is observed during early stage Best disease, but can decline as the disease progresses. In ADVIRC patients, autofluorescence appears normal in the central retina, but is often blocked in the periphery by the characteristic peripheral hyperpigmented ring. 34 Patchy areas of hyperautofluorescence, corresponding to fluid accumulation and small confluent lesions, are typically observed in ARB patients. 21 CNV, a serious complication in bestrophinopathy, can be confirmed using OCT and fluorescein angiography. Despite these modern imaging techniques, genetic testing is generally used to confirm clinically suspected bestrophinopathy and identify novel BEST1 mutations.1,21,28,35,44
Clinical management of bestrophinopathies
There are currently no treatments for the bestrophinopathies; however, complications experienced as a result of disease, can be managed. One common complication is neovascularisation, which, although problematic, has been treated using photodynamic therapy (with the use of verteporfin), 45 photocoagulation or through anti-vascular endothelial growth factor (VEGF) injections, such as Avastin or Bevacizumab.7,46 Interestingly, sufficient VA can be retained in some Best disease patients displaying CNV without any treatment. 14 Complications such as macula edema have been treated using oral acetazolamide with a nepafenac suspension 39 (p. 129), while vitrectomy surgery has been used to repair macular holes. 18 In patients at risk of AACG, monitoring intra-ocular pressure and irido-corneal angle with goniocsopy is recommended. Intraocular pressure can be used to assess the development of glaucoma, which can be controlled with topical drops or treated with prophylactic yttrium aluminium garnet (YAG)-laser iridotomy. 35 Bestrophinopathy patients are at risk of subretinal haemorrhages, they may therefore be advised to avoid contact sport and wear safety glasses at times to circumvent this complication. 47
Bestrophin-1
Bestrophinopathies are caused by mutations in the Bestrophin-1 (BEST1) gene, which maps to a 16 kB region of chromosome 11 (11q13), where it spans 11 exons, producing a 1758 bp transcript expressed exclusively in the RPE, of the adult eye.7,48–50 BEST1 is also expressed in extra-ocular tissues including the trachea, lung, kidney, sperm, colon, and testes, and within astrocytes, neurons, and epithelia of the central nervous system.7,51–53 In addition BEST1 is also expressed in the retina during foetal development. 54 BEST1 is a member of the Bestrophin family, an ancient group of membrane proteins, and has been identified in most metazoan organisms. 55 The coding region, which starts in the second exon, encodes a 68 kD protein, consisting of 585 amino acids 4 with a highly conserved intracellular N-terminal domain containing 4 transmembrane spanning domains and a long diverse cystolic C-terminal domain tail.56–59 Chicken and bacterial proteins have been used to analyse the crystal structure of BEST1, revealing a homo-pentameric organisation comprised of five BEST1 protomers58,59 arranged around a central axis to produce a barrel-shaped pore (Figure 2). The channel protrudes just outside the cell membrane, with the majority of the protein located in the cytosol. In the pentameric structure, Ca2+ clasps within each protomer come together as a belt around the central section of the channel, forming a hydrophobic neck, which is dilated by the binding of cytosolic calcium, allowing the flux of Cl- ions.58,59 Over 250 distinct pathogenic BEST1 mutations have been discovered thus far,48,60 these are thought to affect channel formation, channel function, or protein/channel localisation (Figure 3).

Architecture of the BEST1 channel. (a) The structure of a BEST1 protein unit is divided into four segments, composed of alpha helices represented as S1a-c (red), S2a-h (yellow), S3a-b (blue) and S4a-b (magenta), the transmembrane regions (TM) are indicated. The calcium clasp is represented by a turquoise sphere and the start of the C-terminal tail is coloured green. (b) The BEST1 channel, viewed from the extracellular side, is formed from five BEST1 proteins arranged in a pentameric structure, forming a barrel shaped ion pore (c).

BEST1 disease causing mutations. (a) Annotated BEST1 protein sequence with ClinVar benign/likely benign mutations indicated in green. (b) Potential effects of mutation on BEST1 channel.
The retinal pigment epithelium
Bestrophinopathies primarily affect the RPE, a monolayer of pigmented cells that lies between the neural retina and the choriocapillaris, directly below the cone and rod photoreceptors. RPE cells form tight connections with each other acting as a physical barrier between the retina and the underlying choroid and forming an essential component of the blood-retinal barrier. 61 The RPE apical membrane is scattered with microvilli, which project between the outer segments of the photoreceptors. Despite its simple structure, the RPE performs a number of crucial roles that are needed for photoreceptors to detect light and keep the retina healthy (Figure 4). Disruption in these functions can lead to retinal degeneration and loss of vision.

Crucial roles of retinal pigment epithelium (Adapted from Strauss, 2005 61 ).
The RPE is involved in the phagocytosis and degradation of photoreceptor outer segments (POS). The removal and processing of POS waste prevents the build-up of photo-oxidative by-products, and is crucial for RPE and photoreceptor cell health. 62 Furthermore, essential substances contained within the POS, such as retinal, can be recycled by the RPE, as part of the visual cycle, and returned to photoreceptor cells to enable phototransduction. 61 The RPE is a highly active phagocytic monolayer, engulfing and ingesting around 25,000 outer segment discs per day, with each RPE cell in the macular fovea phagocytosing around 20 POS daily. 63 Changes in cell pH, Ca2+ and ion balance can impact on RPE phagocytosis of POS and affect lysosomal function,61,64 therefore disruption of cell homeostasis can affect the removal of waste, resulting in the build-up of toxic debris within and around cells, ultimately leading to cell atrophy. 65
The RPE maintains a healthy retinal environment and structural integrity by secreting signalling molecules, growth factors, neuroprotective factors and immunosuppressive factors, for example, pigment epithelium-derived factor (PEDF) and VEGF, allowing the RPE to communicate with other tissues. 61 As a crucial component of the blood: retina barrier the RPE is involved in the transepithelial transport of molecules between the retina and the choroid 61 and maintaining the ionic homeostasis of the subretinal environment, transporting water and metabolites (e.g. glucose and lactate), controlling pH and removing waste products. The RPE regulates the buffering of ions in the subretinal space in response to the fast paced, light responsive activity of photoreceptors, maintaining ionic balance and pH. To do this, cells express a number of key pumps, transporter and ion channels at the apical and basal surfaces, including ligand-gated/voltage-gated/Ca2+-activated potassium channels, Na+/K+–ATPase, voltage-dependent/ligand-gated Ca2+ channels, volume-regulated anion channels and Ca2+-activated chloride channels. 66
Role of BEST1
Bestrophinopathies are classed as channelopathies, due to the effects of mutations on the conductance of currents through the cell membrane. BEST1 was first identified as a chloride channel in 2002 after overexpression of exogenous BEST1 in HEK293 cells induced chloride currents that were calcium sensitive. 67 Similarly, studies in human foetal RPE suggest it is an Ca2+ responsive channel, which is required to maintain the RPE transepithelial potential. 68 In addition, structural studies support the hypothesis that BEST1 is a Ca2+ -dependent Cl- channel, controlling the flux of Cl- ions in cells.58,59 However, BEST1 may be involved in a number of processes within epithelial cells. BEST1 interacts with the CaV1.3α1D and CaVβ subunits of L-type voltage-dependent calcium channels,69,70 participating in intracellular Ca2+ signalling and potentially contributing to the generation of the LP.70–72 BEST1 may also regulate intracellular Ca2+, by modulating the release of Ca2+ from endoplasmic reticulum stores, this hypothesis is aided by the finding that some protein can localise away from the membrane.71,73,74 There is also evidence to suggest that BEST1 can act directly as a volume-regulated anion channel, regulating cell volume and homeostasis in RPE cells, 75 a role which would be essential for maintaining cell homeostasis.
Although BEST1 is primarily thought to be a Cl- channel, it is highly permeable to other molecules, such as HCO3-, 76 glutamate 77 and gamma aminobutyric acid (GABA), 78 implying that the channel could potentially serve as a pH sensor/regulator and be involved in neurotransmitter release. In addition, the presence of developmental ocular defects, for example, microcornea and nanophthalmos, in some bestrophinopathy patients, suggests that BEST1 may play a role in normal eye development.16,35 Although protein is only observed in the adult RPE, BEST1 is expressed in human retinal cells during early development. 54 Given that eye development is reliant on correct spatial signalling from neighbouring cells, the expression of mutant BEST1 outside the RPE may affect cell: cell interactions, interfering with normal ocular developmental pathways.79,80
It is widely accepted that the BEST1 channel is activated by the binding of Ca2+, however the protein also contains an ATP binding motif, which may be important in modulating channel function. The binding of ATP can modulate channel activity, resulting in increased currents in the presence of Ca2+. This response is disrupted in iPSC-RPE cells from patients with a mutation in the binding motif, suggesting a relevance to physiological BEST1 function in human cells. 72 ATP is the primary candidate for the substance released during the light peak in electrophysiological recordings, such as the EOG test. The EOG is the defining diagnostic test for bestrophinopathies, with patients generally recording a decrease in the recorded light peak: dark trough ratio. The light peak reflects the increased conductance of Cl- across the RPE basolateral membrane. This is thought to occur in response to an unknown ‘light peak substance’ (LPS), released by photoreceptors after exposure to light, which binds to a receptor on the RPE cells, initiating a cascade that results in depolarisation of the RPE basolateral membrane. Previously, ATP was thought to increase intracellular calcium through purinergic receptors, driving the conductance of chloride Cl- across the basal membrane. 81 However, the direct interaction of ATP with BEST1 within the RPE provides another means to drive depolarisation in response to light.
Model systems for bestrophinopathies
A number of animal models have been used to investigate BEST1 in RPE cells, including rats, mice, and dogs. The rat model of Best disease, created by overexpressing mutated forms of BEST1, localises the protein in the RPE basolateral membrane and displays electrophysiological findings typical of human disease, yet no ocular disease phenotype was observed. 82 Similarly in a BEST1-/- knock out mouse model,5,83 although a reduced light peak, reminiscent of Best disease, was observed, no ocular phenotypes were reported. Knock-in mice expressing mutant forms of BEST1 may represent better rodent models as the reduced electrophysiological responses are also accompanied by lipid accumulation, and retinal detachments.11,84 There may be a limit to how much rodent models can contribute to our knowledge of bestrophinopathies as these animals do not possess a macula and therefore might not develop the equivalent retinal lesions seen in human macular diseases. 85
Although dogs do not have a macula, there is a region in the retina called the area centralis, populated with a higher density of cones and free from large blood vessels. Within this region is a foveal-like region, susceptible to a canine form of recessive bestrophinopathy, 86 termed canine multifocal retinopathy (CMR). CMR affects a number of canine breeds and is caused by mutations in cBEST1, making the dog a naturally occurring animal model of bestrophinopathy.87,88 In these animals, the area centralis is affected by vitelliform lesions typical of Best disease, with focal detachment between the RPE and the neural retina also noted 89 or, more typically, multiple retinal lesions, reminiscent of ARB.21,90 The CMR model is aiding current knowledge of disease progression through lesions, pseudohypopyon and atrophy stages. Loss of RPE apical microvilli leading to microdetachment of the retina is thought to be the earliest features of CMR, indicating an RPE-photoreceptor disease interface for bestrophinopathy.91,92
Much of the early work examining BEST1 in human cells involved overexpression of the protein in the kidney epithelial cell lines, HEK-293 or MDCKII.67,93 Yet, RPE cells have a unique polarisation signature, independent of E-Cadherin, where sorting of proteins to the apical and basement membrane for example, Na+ /K+ ATPase and monocarboxylate transporter 1, is reversed compared to other epithelial cells. 94 Wild type (WT) BEST1 does not localise to the membrane of HEK-293 37 and MDCKII already express endogenous ion channels that could impact on electrophysiological recordings. In addition, overexpression studies in foetal RPE have provided valuable insights into the oligomerisation of BEST1, its role as an anion channel and its involvement in regulation of transepithelial resistance.68,95 However, the availability of foetal RPE tissue limits its use as a common model system.
The advent of induced pluripotent stem cell (iPSC) technology has transformed the modelling of bestrophinopathies in vitro (Figure 5). Reprogramming of human somatic cells to a state of pluripotency using embryonic transcription factors has enabled researchers to derive RPE from patients skin or blood sample, providing a cell model which contains an individual patients unique genetic makeup.96–98 iPSC-derived RPE have a pigmented, cobblestone-like epithelial morphology, replicate many of the functions of RPE and express BEST1 in the basolateral membrane, allowing the investigation of inherited ophthalmic disorders within a disease-in-a-cell system.

Generation of patient derived iPSC-RPE as a human disease model system for the study of bestrophinopathies and development novel forms of treatments for patients.
Disease modelling using iPSC-RPE has replicated many of the features of bestrophinopathies, including reduced channel activity, defects in POS phagocytosis lysosome defects, accumulation of lipofuscin and reduced net fluid transport.40,54,99–102 iPSC-RPE studies have also provided more evidence suggesting BEST1 can function as a voltage gated anion channel, 5 regulate calcium signalling 6 and be regulated by ATP. 72 Cellular studies have identified distinct pathological features that can be used to distinguish between the effects of different mutations 102 and different bestrophinopathies for example, anion transport is increased in ADVIRC cells and decreased in Best disease cells compared to controls. 100 Disease-in-a-dish modelling using iPSC-RPE may yet reveal more about the nature of BEST1 mutations. However, the power of these cells is the ability to investigate new therapeutics by performing drug/compound screens and developing the next generation of personalised genomic medicines.
Therapies for bestrophinopathies
Ophthalmology has been at the forefront of developing the new era of genomic and regenerative medicine approaches, including gene therapy, CRISPR genome editing, antisense oligonucleotide and stem cell therapies. Rapid clinical advances for retinal degeneration have been enabled by the accessibility and small size (6 mm) of the macula, ocular immune privilege, real-time ocular imaging, visual function testing, and the availability of two eyes in a patient–allowing one to serve as an untreated. Currently there are no curative therapies for bestrophinopathies, therefore research is focusing on developing a number of clinical treatments.
Gene therapy
Gene therapy involves introducing exogenous genetic material, that is, a working copy of the BEST1 gene, into the cells of a host in an attempt to treat the underlying cause of a disease. This approach is predicted to be optimal in loss of function mutations, where levels of functional protein are low or absent (Figure 6). A number of research groups have provided proof-of-concept approaches treating bestrophinopathy with a gene augmentation approach. Guziewicz and colleagues, have used subretinal injections of BEST1 adeno-associated virus (AAV)-mediated augmentation gene therapy to reverse the early retinal microdetachments and lesions seen in canine models of ARB (CMR). Improvements were maintained for at least 23 months and no reports of retinal toxicity noted up to 6 weeks post-injection. This approach also preserved the cytoarchitecture of the RPE–Photoreceptor interface and normalised the otherwise diseased hyperthick outer nuclear layer, providing proof of principle that gene therapy may be a viable treatment for patients with CMR-like ARB. 88

Gene therapy approaches for bestrophinopathies can be tested in patient iPSC-RPE cells in a trial in a dish scenario. This approach could be used as a screen to identify mutations responsive to the treatment prior to clinical application in the patient.
IPSC-RPE are also being used to assess the feasibility of BEST1 gene therapy in human cells (Figure 6). Initial studies, using iPSC-RPE from patients with ARB show that gene therapy delivered using baculovirus rescues the Ca2+ -dependent Cl- channel deficiencies observed in cells from patients with recessive disease. 103 Surprisingly, supplemental gene therapy may also be a potential treatment for dominant bestrophinopathies. Ji and colleagues examined iPSC-RPE cells derived from six patients with Best disease, where dominant BEST1 mutations affected the subcellular localisation or function of BEST1. Here they found that treatment of patient iPSC-RPE with BEST1-AAV restored deficiencies in Ca2+ -dependent chloride channel activity, with the viral BEST1 protein localising to the plasma membrane. 102 Despite these successes and exciting proof-of-concepts for recessive and dominant disease, gene therapy may not be a suitable treatment for all BEST1 mutations. Gene augmentation using lentiviral BEST1 was able to restore Ca2+-activated Cl- channel in activity and phagocytosis of outer segments in 3 of 4 patient lines tested, but one cell line, with the Ala146Lys mutation, did not respond to the treatment. This may be due to dominant effect of the mutated protein on the supplemental WT BEST1 protein. 104 Although gene therapy for BEST1 is still in its infancy, early indications in these preclinical models suggest it may be a viable option for recessive disease and, in some cases, dominant disease with responsive mutations, highlighting the need for careful screening of potential gene therapy candidates.
Genome editing
Individual mutations within the BEST1 gene can have different consequences on the protein, affecting its localisation, the formation of the BEST1 pentametric channel formation or channel function. The interaction of the dominant protein with the supplemental protein may still affect individual patients’ responses to gene therapy. In these cases, alternative options need to be investigated. Genome editing provides a therapeutic means to get to the heart of the problem, changing the mutated DNA sequence or switching off a faulty gene. The CRISPR-Cas9 (Clustered Regularly Interspaced short palindromic repeats with CRISPR associated protein 9) system is currently the most popular, efficient and adaptable method to induce permanent changes in cellular DNA. 105
Initially described as a bacterial defence mechanism to identify and inactivate invading viral DNA, CRISPR-Cas9 is now one of the most promising means to treat inherited diseases (31). CRISPR-Cas9 is an RNA guided endonuclease that can be directed to cut double stranded DNA at a specific site upstream of a short protospacer adjacent motif (PAM). The cut site can be determined by providing a complementary single guide RNA (sgRNA), which directs Cas9 to produce a double-strand break at a precise sequence (Figure 7(a)). At this point the cell activates mechanisms to repair the DNA using either homology directed repair (HDR) or non-homologous end joining (NHEJ), which provides opportunities to edit the gene. 106
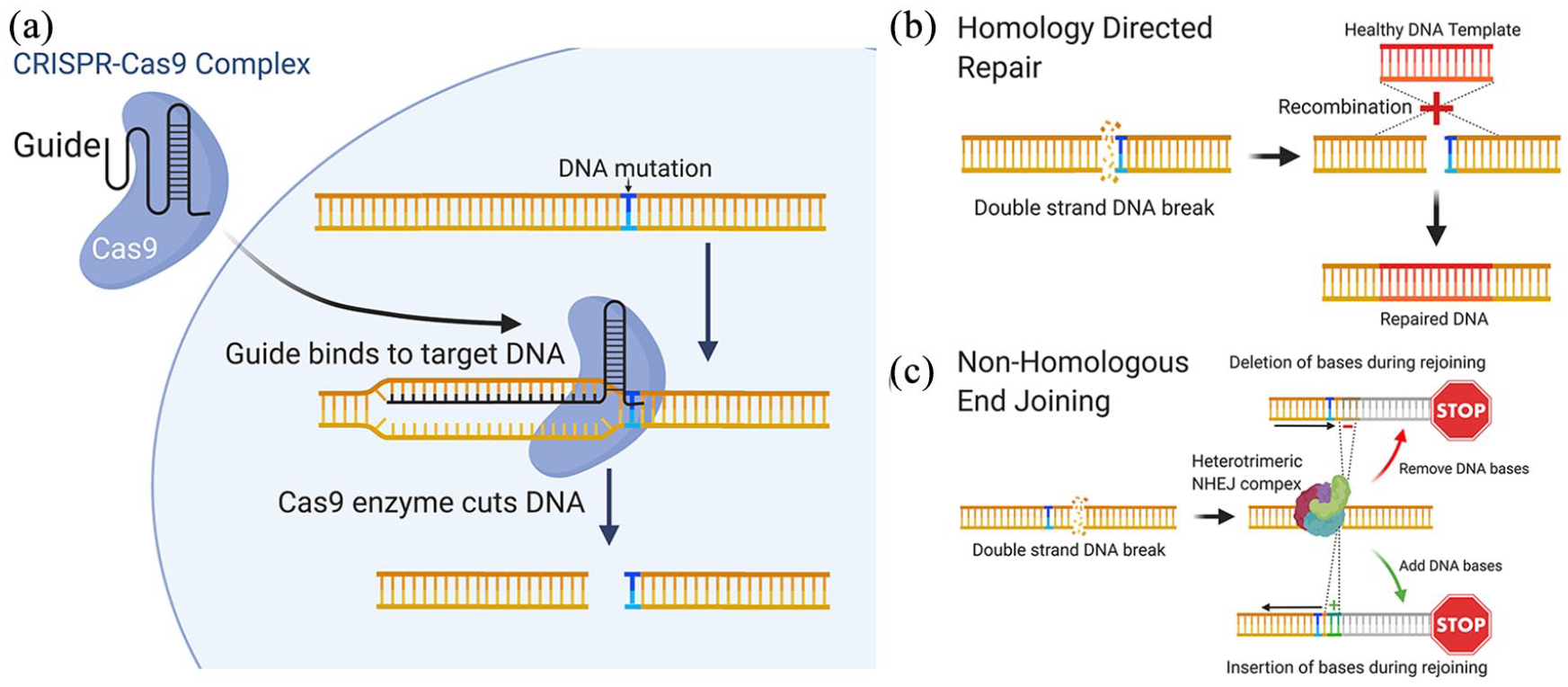
CRISPR genome editing can be used to treat patient mutations at the molecular level. (a) The CRISPR-Cas9 complex is used to create a double strand DNA break in close proximity to the patient. (b) A corrected DNA template can be incorporated into the sequence using homology directed repair, alternatively and (c) non-homologous end joining can be used to create insertions/deletions, resulting in frame-shifts that disrupt the target gene.
Homology-directed repair (HDR) is a critical DNA repair mechanism, commonly active during meiosis, requiring the presence of a homologous DNA for example, a sister chromatid. For genome editing, an sgRNA targeting a mutation site can be introduced into the cell alongside a homologous WT DNA template, which is then incorporated into the gene during the repair, replacing and correcting the mutated DNA region (Figure 7(b)). However due to the practical limitations of HDR, the probability of donor DNA being used as a template in non-dividing somatic cells, like the RPE, is low, therefore HDR repair editing may not be efficient in an RPE cell in situ. An alternative and simpler method of editing uses NHEJ, a common cellular mechanism that joins the two ends of double-stranded DNA breaks back together. This method is highly prone to errors, resulting in indel creation and subsequent transcriptional frameshifts, which lead to degradation of the transcript (Figure 7(c)). NHEJ can therefore be a useful way of switching off a gene in somatic cells, and could be used to target autosomal dominant diseases by directing Cas9 to target specific gene mutation sites on the dominant mutated allele.
Recently, Sinha and colleagues 104 demonstrated the first proof-of-concept approach for gene editing in Best disease using NHEJ, with an average frame shift efficiency of 96%. CRISPR/Cas9 treatment of patient derived iPSC-RPE improved BEST1 channel activity and rhodopsin degradation. However, a major concern was the presence of off-target editing effects, which could affect the expression of other key genes in edited cells. A number of challenges remain before gene editing can be widely used in clinic, including the delivery of the gene editing system to target cells, efficiency of the therapy, utilisation of repair mechanisms in RPE and retinal cells, and prevention of off-target effects.107,108
Pharmacological approaches
Although a great deal of attention is being placed on using genomic medicine to treat inherited diseases, pharmacological approaches may be valid for bestrophinopathies.109,110 Singh and colleagues, 111 investigated whether modulating proteolytic machinery using valproic acid, a histone deacetylase inhibitor, in combination with rapamycin, an inducer of autophagy, could rescue POS processing defects observed in bestrophinopathies. This combinatory treatment increased the rate of POS degradation and reduced the build-up of autofluorescence in patient derived iPSC-RPE, suggesting a link between POS handling and proteolysis in RPE. Furthermore, this treatment also delayed disease progression in a canine model of ARB.
Recent interest has also turned to the use of molecular chaperones and proteasome inhibitors as therapeutics for bestrophinopathies. The proteasome inhibitor, bortezomib and chemical chaperone, 4-phenylbutyrate (4PBA) have been used in combination to guide correct trafficking of exogenous mutant BEST1 to the plasma membrane in MDCKII cells. This combination can also rescue channel activity defects in HEK293 cells expressing inducible forms of mutant BEST1. Similarly, 4PBA and its analogue, 2-naphthoxyacetic acid, were able to increase BEST1 protein expression in iPSC-RPE from Best disease and ARB patients and restore channel function in HEK293 cells expressing mutated forms of BEST1. 112
Conclusion
Bestrophinopathies are distinct retinal dystrophies with varying clinical heterogeneity and penetrance that typically lead to central vision loss at an early age and have a huge impact on the daily lives of patients. The gene responsible for these diseases, BEST1, is a highly conserved anion channel, yet there are few animal models available to fully understand its role in the development of disease, and although BEST1 expression is widespread throughout the body, it is still unclear why BEST1 mutations manifest in vision loss only. Patient derived iPSC-RPE are a crucial disease-in-a-dish model system that have enabled a greater understanding of BEST1 function and its role in human disease. These cells will be important in revealing the role of individual mutations in the development of distinct bestrophinopathies and could help to interrogate the heterogeneity of disease, by identifying potential genetic modifiers of disease in families. In the future, iPSC may also help gain valuable insight into the importance of the RPE: retinal interface in bestrophinopathies, through the culture of retinal organoids. The importance of iPSC-RPE can be fully appreciated in their use to develop and test potential genomic therapies for bestrophinopathies. Current research suggests that, although there may not be a common approach to treat all bestrophinopathies, a range of options could be available for patients in the future, providing permanent treatments for these inherited diseases.
Footnotes
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors gratefully acknowledge funding the Macular Society, the Uren Foundation and the London Project to Cure Blindness.