
Abstract
Inherited metabolic disorders are a large group of rare disorders affecting normal biochemical pathways. Many metabolic disorders can present with symptoms affecting the eye, and eye disorders can evolve later in the natural history of an already diagnosed metabolic disorder. The ophthalmic involvement can be very varied affecting any part of the eye, including abnormalities of cornea, lens dislocation and cataracts, retina and the distal optic pathway, and extraocular muscles. Awareness of inherited metabolic disorders is important to facilitate early diagnosis and in some cases instigate early treatment if a patient presents with eye involvement suggestive of a metabolic disorder. Ophthalmological interventions are also an important component of the multisystem holistic approach to treating patients with metabolic disorders.
Many inherited metabolic disorders (IMD) can have ophthalmological effects, including disorders of any part of the eye, extraocular muscles and visual pathways. Awareness of the potential ophthalmic effects of IMD is important for any clinician including neonatologists, paediatricians and ophthalmologists who may encounter patients whose initial presentation or subsequent disease-course may be with the eye-related feature of a specific IMD. This brief review aims to offer a summary of eye involvement in the different IMD to assist in the earlier recognition and diagnosis of IMD that can facilitate earlier treatment.
Brief introduction/ overview of principles of IMD
The IMD encompass a large group of several hundred individually rare genetic disorders that each affect a specific chemical reaction within the myriad normal biochemical pathways. 1 The majority are caused by absence or deficiency of a specific enzyme that catalyses a step in the biochemical pathway, with subsequent accumulation of the reaction’s substrate which can be directly toxic (such as ammonia in the urea cycle defects), conversion of the substrate via an alternative pathway to a toxic substance (such as the production of succinylacetone in tyrosinaemia type 1), or deficiency of the reaction’s product (such as glucose in the glycogen storage disorder, resulting in hypoglycaemia). Some IMD are due to absence or malfunction of a trans-membrane transporter, and others are caused by dysfunction of whole organelles (such as the peroxisomal biogenesis defects). While some IMD are relatively benign and easy to manage, many
cause multisystem dysfunction and are associated with progressive deterioration over time and are life-limiting. IMD can present with clinical features affecting any or multiple organ systems, including the eye and visual pathways.
Some IMD have specific treatments available in the form of specialised dietetic management, vitamin or co-factor supplementation, recombinant enzyme replacement therapy, or in some situations haematopoietic stem cell or solid organ transplantation. Increasingly gene therapy approaches are also being applied to the treatment of IMDs. Early identification of an IMD can facilitate treatment where available, and important genetic counselling for the wider family.
The majority of IMD are inherited in an autosomal recessive manner, where both parents are unaffected heterozygous carriers of the disorder and the child is only affected as they have inherited both the maternal and paternal mutated allele. In this situation, the recurrence risk for any future children is 1:4 (25%). Some IMD are inherited as autosomal dominant traits, while a small number are X-linked. The mitochondrial disorders can be inherited as autosomal recessive, dominant, or X-linked conditions due to mutations in a nuclear gene, but also inherited matrilineally (i.e. just from the maternal line) if caused by a mutation in the small 16.5 kb ring of mitochondrial DNA containing 37 genes present in the mitochondria. 2
Eye involvement in IMDs
In some situations, eye involvement can be the primary presenting feature of an IMD that can lead to a diagnosis, for example, the identification of neonatal cataracts can be the first sign of disorders of galactose metabolism, while an incidental finding of cornea verticallata (vortex/whorl keratopathy) at a routine optician check can lead to the diagnosis of the lysosomal storage disorder (LSD) Fabry disease. Detection of a corneal cherry red spot is also a ‘red flag’ for a potential LSD. Awareness of the potential for an IMD in such clinical situations is important in facilitating prompt diagnosis and treatment.
Many IMD have ophthalmic involvement as an expected complication or sequela, and these patients will be referred to ophthalmology services to facilitate appropriate monitoring and treatment, and in this situation it is important that knowledge of the natural history and expected evolution of the ophthalmological component of the IMD guides the clinical surveillance.
IMD can affect any part of the visual pathway, from cerebral visual cortex dysfunction in many IMD with progressive central nervous system degeneration, to optic neuropathy, retinopathy, lens opacification and cataracts, corneal clouding and various other keratopathies, and abnormalities of eye movements if the extraocular muscles are affected or if the central optic motor nuclei are dysfunctional. It is important that an IMD is considered in the differential diagnosis of the eye condition, alongside common acquired or other non-metabolic genetic causes.
General diagnostic approach
Identification of an IMD requires a low threshold of clinical suspicion. Within the clinical history, evidence of extra-ocular features should be sought to determine if the presenting eye signs or symptoms are in the context of a multisystem disease. The age of the child is important; a neonate with failure to thrive, recurrent vomiting, prolonged jaundice or other developmental concerns may have an IMD. The family history is important, especially other affected siblings or relatives, unexplained neonatal or childhood deaths, recurrent miscarriages. Enquiring about parental consanguinity is important as many of the IMD are inherited in an autosomal recessive manner, although these conditions still occur in children of non-consanguineous parents. Subsequent dietary preferences, abnormalities of normal childhood developmental progress (especially developmental regression, i.e. loss of developmental milestones previously achieved), detection of hepato/splenomegaly or dysmorphic features can also allude to an IMD. Some children will have been reviewed by a number of specialists addressing individual manifestations of an IMD without the diagnosis being made, for example, a child with a mucopolysaccharidosis (MPS) disorder may have seen ENT surgeons for recurrent otitis media or obstructive sleep apnoea, general surgeons for inguinal or umbilical hernia, and community paediatrician for developmental delay, as well as ophthalmology services due to the development of corneal clouding, before the diagnosis is reached. Some children may have imaging studies undertaken, such as brain magnetic resonance imaging (MRI), and these can also reveal diagnostic information regarding IMD if a specific pattern such as a leukodystrophy is identified. 3
In planning specific investigations it is important to review any prior specific biochemical/ metabolic tests that have been undertaken, and to then ensure that key diagnostic samples are collected determined by the specific ophthalmological problem detected. Metabolic testing will include general biochemistry (e.g. renal and liver function tests, cholesterol, creatine kinase [CK], uric acid), and specific metabolic testing on blood and urine samples (such as plasma amino acids, urine organic acids, urine glycosaminoglycans). In many situations, the diagnosis is then confirmed with specific enzyme assays which are done in blood samples, or occasionally in tissues such as cultured skin fibroblasts, or muscle/liver biopsies. 4 Increasingly molecular genetic testing has a first-line role in diagnostic pathways, as well as the more typical role in confirmation of a biochemical diagnosis. For example, a number of next generation sequencing panels for genes causing cataracts may be employed that include genes causing IMDs. Furthermore, whole genome sequencing initiatives (such as the UK 100,000 genome project) may identify a specific but unexpected IMD, and in this situation biochemical assays may be required to substantiate and evaluate the genomic variants to determine pathogenicity.
Early discussion with a metabolic specialist can help with planning key investigations, and in some instances with instigating pre-emptive treatment while investigation results are sought such as appropriate galactose/lactose-free feeds in a neonate at risk of galactosaemia.
Specific eye findings
Table 1 provides a summary of the metabolic differential diagnosis for each eye finding, together with specific extra-ocular features, key diagnostic tests and specific treatments, and key literature references.
Summary of metabolic differentials, extra-ocular features, diagnostic tests and treatments for specific ophthalmic findings.
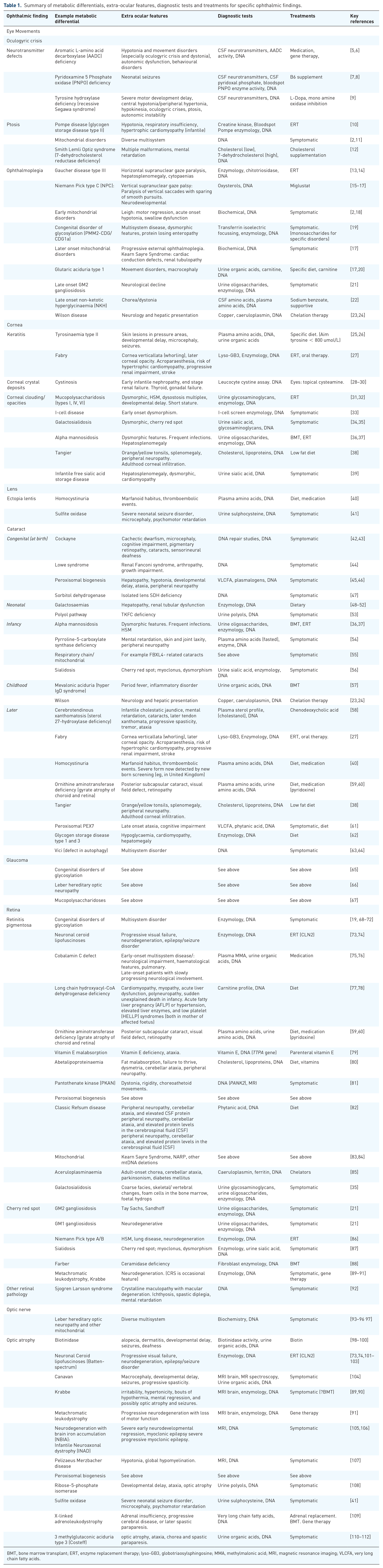
BMT, bone marrow transplant; ERT, enzyme replacement therapy; lyso-GB3, globotriaosylsphingosine; MMA, methylmalonic acid; MRI, magnetic resonance imaging; VLCFA, very long chain fatty acids.
Summary of investigations.

Eye movement disorders
Oculogyric crisis
Neonatal or early-onset oculogyric crisis may be due to a defect in neurotransmitter metabolism, and will usually be seen as part of a severe neurological phenotype with hypertonia and/or seizures; these can occur in disorders of vitamin B6 metabolism that are treatable with oral vitamin B6 supplementation. Diagnosis usually requires cerebrospinal fluid analysis of neurotransmitters together with molecular genetic testing. Treatment will depend on the specific diagnosis, and may include neurotransmitter replacement or specific blockade therapies.
Ophthalmoplegia
The differential diagnosis of new onset strabismus in early childhood is broad including simple refractive errors, space occupying lesions, but also including IMD. Strabismus is a feature of the commonest form of congenital disorder of glycosylation (CDG), phosphomannomutase 2 (PMM2-CDG) (previously known as CDG-Ia). These disorders impair the normal process of post-translational modification of proteins, and the abnormal sugar side chain (glycosylation) results in abnormal function of the mature protein. Ocular involvement in this multisystem disorder can also include nystagmus with or without visual impairment, and other subtypes of CDG also have eye involvement. Extraocular manifestations are expected including dysmorphic features, protein-losing enteropathy, pericardial effusions. 15
Early-onset mitochondrial disorders including those presenting with Leigh and Leigh-like syndrome can present with new-onset squint together with other loss of previously acquired skills often after a minor febrile illness; imaging studies together with biochemical testing are the first steps in the diagnostic workup. Mitochondrial disorders can also present later in life with primary ocular motor defects, including progressive external ophthalmoplegia as either an isolated feature, or as part of a multisystem mitochondrial defect such as Kearn Sayre Syndrome. Importantly, some mitochondrial disorders such as autosomal recessive polymerase gamma (POLG)-related disease can present in early childhood with severe disease, where parents who are heterozygous carriers may be at risk of later onset autosomal dominant disease manifestations with particular mutations, warranting careful genetic counselling.
Some LSDs have specific eye movement abnormalities. Gaucher disease can be associated with a horizontal supranuclear gaze palsy that can be subtle and detected only with careful rotational movement assessments. 13 In this situation, the detection of eye movement abnormalities can help differentiate patients with type III (chronic neuronopathic Gaucher disease) from the non-neuronopathic type 1 disease, guiding therapeutic approaches.
Niemann Pick type C (NPC) is a cholesterol trafficking LSD; the age of presentation is very wide, but the typical eye movement defect is of a vertical supranuclear gaze palsy with paralysis of vertical saccades with sparing of smooth pursuits in the initial stages. This is seen in conjunction with developmental regression and splenomegaly in the early infantile forms, but may be an isolated feature initially in later onset disease. Treatment options include substrate reduction therapy with miglustat that can ameliorate the disease course.
Ptosis
Ptosis can be seen in a wide range of myopathies and myasthenic syndromes. Of the IMD, mitochondrial disorders can present with ptosis that is often worse with fatigue. Of the metabolic myopathies, Pompe disease (glycogen storage disease type II) may have progressive ptosis together with generalised motor and respiratory myopathy, and in infants also hypertrophic cardiomyopathy. Treatment with enzyme replacement therapy is available.
Corneal pathology
Keratopathy
Tyrosinaemia type II is a rare amino acid disorder that causes a painful keratitis, resulting in photophobia, lacrimation and burning pain. It is also associated with skin lesions particularly in pressure areas, and also neurodevelopmental delay with microcephaly and seizures in some patients. Slit lamp examination may reveal bilateral herpetic-like lesions with neovascularisation, which untreated can result in corneal scarring. Diagnosis is by identifying very high plasma tyrosine levels and excluding other subtypes of tyrosinaemia. Treatment with dietary measures to decrease the tyrosine level is effective.25,26
Fabry disease is an X-linked LSD that causes multisystem disease including progressive renal impairment, hypertrophic cardiomyopathy and strokes, although the phenotype is very variable. Females can be affected. Children present with early symptoms of painful acroparaesthesia. Patients may be detected incidentally in a pre-symptomatic stage by the finding of cornea verticallata (corneal whorling); at later stages corneal opacification manifests. Specific therapy is available with intravenous enzyme replacement, and oral disease modifying drugs, and so early diagnosis is important. 27
Corneal clouding/opacification
Corneal clouding and opacification is a feature of several IMD, in particular some of the progressive LSD conditions, 113 and these should be considered alongside other acquired causes of corneal opacification especially when bilateral.
Cystinosis is a LSD caused by a defect in the lysosomal cysteine exporter, and in its severe form causes an early infantile nephropathy with end stage renal failure. Eye involvement is usually at a later stage, and thyroid and gonadal pathology is also expected. There are milder forms isolated to ocular involvement. Cystine deposits accumulate within the stroma of the cornea, iris, lens and retina, and manifest as photophobia with visual defect. Systemic treatments as well as topical cysteamine are used.28–30
Other LSD that present with corneal clouding included the MPS (e.g. MPS I Hurler syndrome, MPS IV (Morquio), MPS VI (Maroteaux Lamy), but not the X-linked MPS II Hunter syndrome),31,32 sialic-acid-related LSD (galactosialidosis, sialic acid storage diseases), and other oligosaccharidoses such as alpha-mannosidosis. These are all associated with multisystem disease and progressive dysmorphic features; patients may have seen a number of specialists before a diagnosis is reached, emphasising the need to consider these disorders. In some attenuated forms of MPS, the ocular manifestations may be the initial presenting feature. 114 Surgical intervention to address the corneal clouding may be necessary, as the response of the corneal clouding to intravenous enzyme replacement therapies is very variable; corneal transplant has been successfully applied in this situation with very important benefits for the patients.
Lipid-associated disorders can also present with corneal pathology. Tangier disease is a rare disorder affecting cholesterol efflux, associated with orange/yellow tonsils, splenomegaly, peripheral neuropathy and the later development of corneal infiltration. It is treated with a low fat diet. 38 Corneal arcus is seen in patients with hypercholesterolaemia, and if this is seen in young patients may be a sign of the more severe forms of hypercholesterolaemia such as homozygous familial hypercholesterolaemia, where urgent treatment to avoid very early coronary artery morbidity and mortality is required.
Lens pathology
Ectopia lentis
Dislocation of the optic lens has a range of causes including Marfan syndrome, and the Weill-Marchesani syndromes115,116 while metabolic causes include homocystinuria, which is classically associated with downwards-dislocation where this may be the initial presenting feature, and the severe disorder sulfite oxidase deficiency that is associated with early onset seizures. Appropriate metabolic screening should be undertaken in patients with unexplained lens dislocation. While homocystinuria is now part of routine newborn screening in many countries including the United Kingdom, patients born prior to screening may still present symptomatically.
Cataract
The development of paediatric cataract always requires investigation to determine the aetiology. In many situations, this is a primary genetic disorder affecting the transcription factors such as Pax6, and c-Maf that have pivotal roles in the development of the lens and its polarity. 117 A number of IMD can also present with cataracts, and the age that the cataract develops can help in the differential diagnosis.
Truly congenital cataract, that is, those present already at birth, may suggest conditions including Cockayne syndrome which is a DNA repair defect associated with severe short stature, retinitis pigmentosa (RP), sensorineural deafness and early mortality;42,43 Lowe syndrome, an X-linked disorder comprising cataracts, proximal renal tubular dysfunction and intellectual impairment due to mutations in the OCRL gene encoding OCRL-1 which is an inositol polyphosphate 5-phosphatase that has roles in endocytic trafficking; 44 and isolated sorbitol dehydrogenase deficiency specifically affecting the lens. 47
An important category of IMD causing cataracts are the peroxisomal disorders. 45 Peroxisomes are intracellular organelles with a wide range of biochemical functions including beta-oxidation of very long chain fatty acids, alpha-oxidation of phytanic acid, and bile acid and plasmalogen synthesis. Peroxisomal defects include single enzyme defects (such as x-linked adrenoleukodystrophy) or defects affecting the biogenesis of the whole peroxisome (the Zellweger spectrum disorder [ZSD] peroxisomal biogenesis defects). Patients with ZSD can have congenital cataracts; there are numerous genes involved in peroxisomal biogenesis, and several have been associated with ocular disease including PEX2, PEX11B, PEX10, PEX12 and PEX16 (see reference Steinberg and colleagues 45 for summary). These may even be detected antenatally. 118 The severe forms of ZSD present in the neonatal period with severe hypotonia, enlarged fontanelle, hepatopathy and significantly shortened life expectancy, but those at the milder end of the spectrum can present much later. Cataract can be the presenting feature, and careful diagnostic work up is required, especially as the classical biochemical biomarker (very long chain fatty acids) may not show consistent abnormalities, such as in patients with PEX11B. 46 Later onset cataracts in adulthood can also be due to peroxisomal biogenesis defects, for example, in PEX7-related late onset ataxia and cognitive impairment with cataracts. 61 Biochemical testing for peroxisomal defects includes assays of the very long chain fatty acids and other products of peroxisomal function; many of the biogenesis genes are also incorporated into gene panels for investigation of cataracts.
Neonatal onset cataracts are a feature of the galactose metabolism disorders. Galactose is derived from the disaccharide lactose, the main carbohydrate in mammalian milk. Lactose is hydrolysed by intestinal lactase generating galactose and glucose which are then absorbed across the brush border by the SGLT1 co-transporter. Galactose is phosphorylated to galactose-1-phosphate by the galactokinase (GALK), and is then converted to uridine diphosphogalactose (UDPgalactose) by galactose-1-phosphate uridyltransferase (GALT or ‘Gal-1-put’). UDPgalactose is an important source of galactose for the generation of complex glycoconjugates such as glycoproteins and glycosaminoglycans. Galactose can also be converted to galactitol via aldose reductase; and UDPglucose can be interconverted to UDPgalactose by UDPgalactose-4-epimerase (GALE).
Disorders of galactose metabolism are associated with defects in any of these enzymes (GALT/Gal-1-put, GALK, epimerase/GALE). Cataract formation in all of the disorders occurs due to the accumulation of galactitol in the crystalline lens. Classical galactosaemia is caused by GALT deficiency, and is associated with very early oil drop cataract; this may resolve spontaneously if treatment with galactose-free diet is introduced promptly, but the cataract may mature and require surgical correction if left untreated (see Figure 1).48–50 Progressive liver dysfunction with jaundice and coagulopathy, renal and cerebral disease manifest in the neonatal period in children with classical galactosaemia, and urgent treatment is required to avert death. Epimerase (GALE) deficiency can present with severe neonatal disease, or can remain asymptomatic if there is residual enzyme activity; juvenile cataracts may be a presenting feature. 51 In galactokinase deficiency cataracts are the only clinical feature seen, due to the osmotic effect of galactitol causing swelling of fibres within the lens. Liver disease is not seen, and treatment with galactose restriction can lead to resolution of the cataracts if they are not established. 52

Atypical presentation of classical galactosaemia [from JED clinical archive].
Testing for the galactosaemias includes erythrocyte enzyme assays, which are therefore not valid after a recent red cell blood transfusion. Erythrocyte galactose-1-phosphate levels are measured. In some situations (eg if a sick neonate has received a red cell transfusion) testing Gal-1-put activity in both parents can establish if they have heterozygous enzyme activity levels. Molecular genetic testing will help confirm the diagnosis. In all situations where galactosaemia is suspected, immediate treatment with dietary galactose restriction is mandatory until testing is completed to confirm or refute the diagnosis.
Next generation sequencing molecular genetic technologies have been employed in the investigation of children with idiopathic cataracts, for example, identifying a novel disorder also affecting the polyol pathways linked with galactitol accumulation. 53
Cataracts appearing during infancy and childhood may be due to many different IMD. The LSD conditions including alpha mannosidosis, sialidosis and Fabry disease all are associated with cataracts, and would usually be seen in the context of multisystem disease. A recently described primary disorder of autophagy (the process of internal cellular housekeeping), Vici syndrome, also includes cataracts in its constellation of symptoms.63,64
Juvenile/adult onset cataracts are seen in cerebrotendinous xanthomatosis (CTX), associated with progressive neurological abnormalities and tendon xanthomata, and Wilson disease can also present with ‘sunflower cataracts’ as well as the classical Kaiser-Fleischer rings.23,24
Late (adult) onset cataracts may be seen in several IMD, including in gyrate atrophy of choroid and retina due to ornithine aminotransferase deficiency where posterior capsule cataracts are associated with visual field defects and retinopathy; affected patients may benefit from dietary measures and some oral medications have been trialled. These patients may have had biochemical abnormalities in the neonatal period that can include transient hyperammonaemia.59,60
Glaucoma
Raised intraocular pressure may be seen as a secondary feature in several IMD with other ocular pathologies, including in the congenital disorders of glycosylation, 65 Leber’s hereditary optic neuropathy 66 and the mucopolysaccharidoses. 67
Retina
Retinitis pigmentosa
Most cases of RP with loss of retinal photoreceptor cells and pigmentary deposits are caused by primary genetic defects, either autosomal dominant or recessive.119,120 However, RP is seen as part of many different IMD.
Primary vitamin E malabsorption, or disorders with secondary vitamin E and A deficiency such as abetalipoproteinaemia, cause RP together with other neurological manifestations of vitamin E deficiency. The progression of eye disease in these conditions can be altered with high dose vitamin supplementation.
The Cobalamin C defect in the vitamin B12 metabolic pathway is associated with RP, together with multisystem disease including neurological, haematological and cardio-pulmonary features in early onset patients, or a milder phenotype with slower progressing neurological disease with visual loss, although the visual defect may progress despite treatment.75,76
Disorders of brain iron accumulation (neurodegeneration with brain iron accumulation [NBIA]) including infantile neuroaxonal dystrophy (INAD) and pantothenate kinase deficiency (PKAN) incorporate retinal pathology,105,106 with pigmentary retinopathy reported in 58% of early-onset PKAN patients and 15% of late-onset patients. 81 Treatments are currently symptomatic only; the diagnosis is often suggested due to specific features on MRI neuroimaging showing iron accumulation in specific brain regions.
The neuronal ceroid lipofuscinoses (Batten-disease spectrum) conditions have a very important ocular component including RP and progressive optic atrophy, with visual loss being a hallmark of many of the subtypes associated with 13 different genes.73,74,101–103 Recently disease modifying treatment for one subtype (CLN2) has been licenced, and gene therapy approaches are being investigated for other subtypes.
Mitochondrial disorders can present with RP with visual failure as a primary feature, for example in neuropathy, ataxia with retinitis pigmentosa (NARP).83,84
Cherry red spot
The retinal ‘cherry red spot’ detected on fundoscopy describes the finding of a deep red circular area temporal to the optic disc at the centre of macula, surrounded by a pale ‘halo’. The pale halo is caused by accumulation of storage material within the retinal cells which lose their normal transparency, while the foveola which does not contain ganglion cells remains transparent and so the choroid vasculature remains visible and generates the ‘red spot’.121,122 It is seen as an acquired phenomenon if there is central retinal artery thrombosis, but in children is usually due to an IMD and its identification can help refine the differential diagnosis considerably in a child with multisystem disease. Specifically several LSDs are associated with the cherry red spot as these conditions result in accumulation of intracellular storage material. These disorders may also have hepatosplenomegaly as in GM1 gangliosidosis, galactosialidosis or Niemann Pick A disease, and dysmorphic facial features may also be evident. Prominent myoclonus or startle response is seen in GM2 gangliosidoses (Tay Sachs and Sandhoff variants). Diagnosis relies on both urinary studies identifying the storage material, and enzyme assays with confirmation with molecular genetic testing. As there is phenotypic overlap, a panel approach to both enzymology and molecular genetic testing is warranted. 4 Treatment is mostly symptomatic, although disease modifying treatments are becoming available for some LSDs, and this together with the importance of genetic counselling for the family highlights the importance of prompt diagnosis.
Optic atrophy
Optic atrophy, i.e. degeneration of the retinal ganglion cells with optic nerve pallor due to deterioration of the optic nerve at any point in its course, can manifest in many IMD. Optic atrophy is an important feature in many of the mitochondrial disorders, including Leber hereditary optic neuropathy (LHON). 97 The ocular features of mitochondrial disorders are very diverse, and include as described previously disorders of ocular motor function as well as visual function.93–96 The diagnosis of mitochondrial disease can be challenging, given the significant heterogeneity in clinical presentation, time course of disease evolution, and the complex genetics of mitochondrial function. 2 Biotinidase deficiency is eminently treatable with simple oral biotin, which can prevent the progression of optic atrophy, and indeed is screened for in many countries as part of newborn screening.98–100
Optic atrophy can be seen in many of the lysosomal disorders including Krabbe and metachromatic leukodystrophy where there is progressive loss of cerebral nervous tissue, the peroxisomal biogenesis disorders and X-linked adrenoleukodystrophy.
Treatments
Treatment approaches to the different IMD are varied depending on the aetiology of the condition. Symptomatic management is the mainstay for many of the disorders. Some IMD have specific dietetic interventions (e.g. galactose restricted diet in the galactosaemias), while others respond to high dose vitamin or cofactor supplementation. For a number of the LSDs, enzyme replacement therapy is available. The impact of these therapies on the ophthalmic manifestations of the disease is very variable, in some situations (e.g. vitamin E replacement in primary vitamin E deficiency or abetalipoproteinaemia, or biotin replacement in biotinidase deficiency) this may prevent or reverse visual impairment, but in others the eye is relatively refractory to therapy.
Specific ophthalmological interventions may be required. For those with refractive error or visual impairment, appropriate provision of glasses or other visual aids will be required. Standard interventions for correction of strabismus may be undertaken, although understanding the natural history of each disorder is important in determining whether a therapy is appropriate. Management of cataract may require lensectomy, although in some situations instigation of appropriate dietetic management may facilitate resolution of the cataract if not established. For some conditions other surgical interventions including corneal grafting to correct corneal clouding has been beneficial, but appropriate investigation to determine retinal function should be undertaken in the surgical planning process as there may be concurrent optic atrophy or visual loss if there has been chronic uncorrected raised intraocular pressure. Certain disorders may lead to specific topical treatments such as cysteamine in cystinosis.
Conclusion
This brief review has summarised some of the many IMD that may present initially, or subsequently, with ophthalmic involvement. Early diagnosis of an IMD may be made if this is considered in the differential diagnosis of the ophthalmic complaint, which may facilitate earlier treatment. Discussion with the metabolic clinical and biochemical services can ensure that appropriate diagnostics are undertaken.