
Abstract
Is it possible that some mitochondrial DNA (mtDNA) mutations enhance the risk of developing a headache disorder while other mutations actually confer a protective effect? Mitochondrial disorders have been linked to migraine but very rarely to cluster headache (CH). The true pathogenesis of CH is unknown but a linkage to cigarette smoking is irrefutable. Leber’s hereditary optic neuropathy is a syndrome of bilateral vision loss that typically manifests in a patient’s 20s and 30s, is male predominant, and its sufferers are heavy smokers and heavy drinkers. Tobacco exposure is so linked to the condition that only smokers appear to develop vision loss while nonsmokers remain unaffected carriers of their mutations. In essence, the Leber’s hereditary optic neuropathy population is the CH population but at present there have been no reported cases of CH in this mitochondrial subgroup. Thus, could the effects of the mtDNA mutations found in Leber’s hereditary optic neuropathy, which involve complex I of the electron transport chain, actually confer a protective effect against the development of CH? This article will delve into this theory.
Introduction
Mitochondrial disorders have been linked to headache conditions, especially migraine. 1 However, there have only been sparse reports about a possible linkage of mitochondrial DNA (mtDNA) mutations and cluster headache (CH). 2,3 Is it possible that some mitochondrial mutations enhance the risk of developing a headache disorder while other mutations actually confer a protective effect? CH is the fiercest of all headache conditions and its underlying pathogenesis is still unknown. What is hypothesized is that tobacco exposure, typically both as a child and as an adult, seems to play a critical role in the initiation of CH in the majority of patients; along with a history of alcohol overuse and head trauma. 4 Interestingly, there is a mitochondrial patient population which uniquely mimics the CH population. Leber’s hereditary optic neuropathy (LHON) is a syndrome of painless, bilateral vision loss. It is male predominant, and its sufferers are typically heavy smokers and heavy drinkers. 5 Around 70% of LHON patients smoke cigarettes, which is approximately the same percentage noted in the CH population. 5,6 Tobacco exposure is so linked to LHON, that only smokers appear to develop vision loss while nonsmokers remain unaffected carriers of their mitochondrial mutations. 7,8 Indeed, if there is a patient population that should develop CH, it is the LHON subgroup; however, there have been no documented cases published in the medical literature. As LHON is rare and CH is uncommon, the lack of identification of CH in the LHON population could just be the consequence of so few individuals with LHON. However, it is conceivable that the mtDNA mutations that cause LHON also create a protective effect against the development of CH, especially in cigarette smokers. This information could help us better understand CH pathogenesis and possibly provide new treatments in the future. The goal of this article is to delve into the possible protective effects of mitochondrial Complex 1 mutations on CH pathogenesis. The following is a theory based on clinical observations, but in which clear scientific data are still needed to prove the hypothesis.
Demographics
The similarities between the LHON and the CH population are uncanny. Both are primarily disorders of men with at least a 3:1 gender ratio. 5,9 Both syndromes typically start in a patient’s late teens or twenties with rare earlier and later onset cases. Both are uniquely tied to a history of cigarette smoking and alcohol use. In reality, the LHON population is the CH population but with no reported cases of CH. As CH occurs in 1–2 of every 1000 individuals and there are at least 35,000 individuals worldwide with LHON, then at least one case if not more than 30 cases of CH should be reported. 5 From the US CH survey, the largest study of CH patients completed in the United States (1134 individuals), of which 73% had a smoking history, there were no reported patients with a diagnosis of LHON. 6 The Internet survey allowed a write in section to provide any additional past and current medical history. From the Mayo Clinic electronic medical record database of almost 9.5 million patients seen over the last 20 years, there were 23 patients with a diagnosis of LHON and 1090 patients with a CH diagnosis. 10 A history of tobacco exposure was noted in 53% of CH patients, while 30% of LHON patients had a smoking history. None of the LHON patients had a comorbid diagnosis of CH. A recent Danish population–based study of LHON did not elicit any significant comorbid migraine history above the general population and CH was not noted. 11
LHON pathogenesis
LHON causes a subacute loss of vision secondary to degeneration of the retinal ganglion cells. The majority (90%) of individuals who develop LHON typically have one of three point mutations involving complex 1 subunit genes of the electron transport chain. These mutations occur at nucleotide positions: 11,778 (most common), 3460, and 14,484. LHON demonstrates incomplete penetrance with about 50% of male and 10% of female carriers developing vision loss. 5 The exact manifestations of the mtDNA mutations found in LHON are still unknown but there appears to be a defect in ATP synthesis, a disruption in glutamate transport and an increased production of reactive oxygen species. 5 Research suggests that environmental triggers are necessary to initiate vision loss in carriers and especially in males. In one of the largest studies of LHON patients (402 individuals) completed, 73% of symptomatic individuals were smokers compared with 55% of unaffected carriers. There was a significantly higher pack year and daily consumption of cigarettes smoked per day history in the affected patients. Smoking in males was associated with a 93% lifetime penetrance of visual loss versus 66% in nonsmoking males and 33% in smoking females (estrogen protective effect?). 7 In addition, almost all LHON patients (>90%) utilized alcohol and the mean weekly consumption was significantly higher in the affected population; however, there was a much weaker relationship to visual loss than with smoking. Head injury was also associated with vision loss.
Why smoking may lead to the conversion of an unaffected LHON carrier to a symptomatic one has been hypothesized. Cigarette smoking in general has been shown to have deleterious effects on mitochondrial activity including a disruption in respiratory function and an increase in reactive oxygen species. 7,12 Specific to LHON, smoking has been shown to significantly reduce mtDNA copy numbers and cause disruption to complexes I and IV of the electron transport chain with a resultant decline in ATP production. 8 Ultimately, it is felt that smokers with LHON have less compensatory responses to their mitochondrial dysfunction and that is why they develop vision loss.
Mitochondrial dysfunction in CH
There are hints from magnetic resonance (MR) spectroscopy studies that CH patients have some evidence of mitochondrial dysfunction. ATP production is altered both during and after a cluster period. 13 A single case report of an mtDNA mutation (mitochondrial tRNA(Leu(UUR)) in a presumed CH patient was reported in 1994, but this mutation was not identified in subsequent studies from CH sufferers in Italy and Germany. 2,14 A second possible single case of CH in a patient with familial chronic progressive external ophthalmoplegia was published. 3 This patient however may not have had CH based on duration of headache pain (7 h) and attacks only occurring for 7 days. There have been no other reports of specific mtDNA mutations noted in CH patients in the literature, suggesting that either CH is not related to mtDNA mutations or these mutations are protective against CH initiation. It is also possible that the recognized mitochondrial respiration issues noted in previous MR spectroscopy studies of CH patients was actually a smoking effect and not CH specific. The smoking history of the enrolled study patients was not reported. 13
Possible mechanisms by which patients with LHON are protected against the development of CH
Lack of “Double Tobacco Exposure”: The author has previously identified that the majority of CH patients have extensive secondary smoke exposure as a child and then a personal smoking history prior to CH onset and that this “double exposure” may be required to develop CH.
4
Further verification of this hypothesis is needed. At present, there have been almost no studies looking at secondary smoke exposure as a child in LHON patients.
7,8
One investigation did state that in a small group of very young onset LHON sufferers (age <10 years), there was not an elevated rate of smoking in family members, but the authors did not specifically note if there was a lack of smoking history in the parents of affected patients.
5
If double exposure is present in subjects with LHON, this may solidify the conclusion that the sequelae of the LHON mtDNA mutations are protective against developing CH. If secondary smoke exposure as a child is absent, this could be one of the main reasons why LHON patients do not develop CH.
Retinohypothalamic tract and melatonin
In LHON, there is a massive loss of retinal ganglion cells (RGCs) secondary to mitochondrial impairment. However, there is an actual sparing of melanopsin or intrinsically photosensitive RGCs (ipRGCs), suggesting a resistance to degeneration of these cells from metabolic insult. 15,16 The ipRGCs help make up the retinohypothalamic tract by directly linking optic impulses to the suprachiasmatic nucleus (SCN). Human circadian clock entrainment to external light and darkness is dependent upon photic transmission from eyes to the SCN via the ipRGCs. The ipRGCs also support the nonimage forming visual functions of the eye, which include the photo-entrainment of circadian rhythms, light-induced suppression of melatonin secretion, and control of the pupillary light reflex. 17 With the preservation of ipRGCs in LHON, there is a stability of the retinohypothalamic tract. 16 This stability however may not be present in CH patients. There appears to be an inability to entrain the circadian rhythm especially at night and recent studies have noted an abnormal pupillary light reflex in CH sufferers. 18
In the retina, pituitary adenylate cyclase–activating polypeptide (PACAP) is expressed exclusively in ipRGCs. 19 PACAP is a modulator of nociception and melatonin synthesis. PACAP levels have been found to be low in between CH bouts and thus may lead to an enhanced susceptibility for further attacks/cycles. 20 Outside of ipRGCs, PACAP is produced in the sphenopalatine ganglion, the otic ganglion, and the trigeminal ganglion; thus preservation of ipRGCs in LHON may or may not provide enough PACAP to protect against CH development. In addition, it has recently been shown in animal models that the late night phase advance of the circadian rhythm is PACAP dependent and that the nonvisual photic regulation of the circadian rhythmicity in the SCN is dependent on PACAP supplied by retinohypothalamic inputs and not from other hypothalamic regions projecting to the SCN. 19 Preservation of ipRGCs in LHON promotes a preserved retinohypothalmic tract and normal production of ipRGC derived PACAP and thus possible protective mechanisms against the initiation of CH.
Moreover, plasma melatonin levels, while being suppressed in CH patients, are actually elevated in LHON sufferers. 4,21 It has been hypothesized that a change in the pattern of light input to LHON patients causes a redistribution and a secondary increased secretion of melatonin. Suppression of melatonin levels may be a needed requirement for CH initiation and thus elevated levels in LHON patients is another possible protective mechanism against CH development. 21 The exact role melatonin plays in CH pathogenesis still needs further study.
An alternative explanation is that blindness alone could be protective against the development of CH, possibly secondary to the alteration in melatonin levels that occurs with progressive vision loss.
22
The inherited juvenile macular degeneration syndromes can lead to vision loss in a patient’s first or second decade of life (demographic overlap with CH) and as of yet there have been no reports of CH in this population.
23
In addition, smoking has been linked as a major risk factor for the development of age-related macular degeneration, which is one of the leading causes of blindness in individuals
Amino acid alterations/metabolomics profile
Several recent studies have looked at the metabolomics profile of LHON patients versus controls. 25,26 The key finding is a significant decrease in the levels of all proteinogenic amino acids including lysine, threonine, aspartate, glycine, asparagine, glutamate, alanine, histidine, phenylalanine, serine, glutamine, tryptophan, tyrosine, leucine, methionine, valine, isoleucine, proline, and arginine. 25,26 Studies of amino acid levels have also been carried out in CH patients. Table 1 denotes the differences between LHON and CH sufferers. 25 –29
Differences between cluster headache and LHON (amino acids and other).
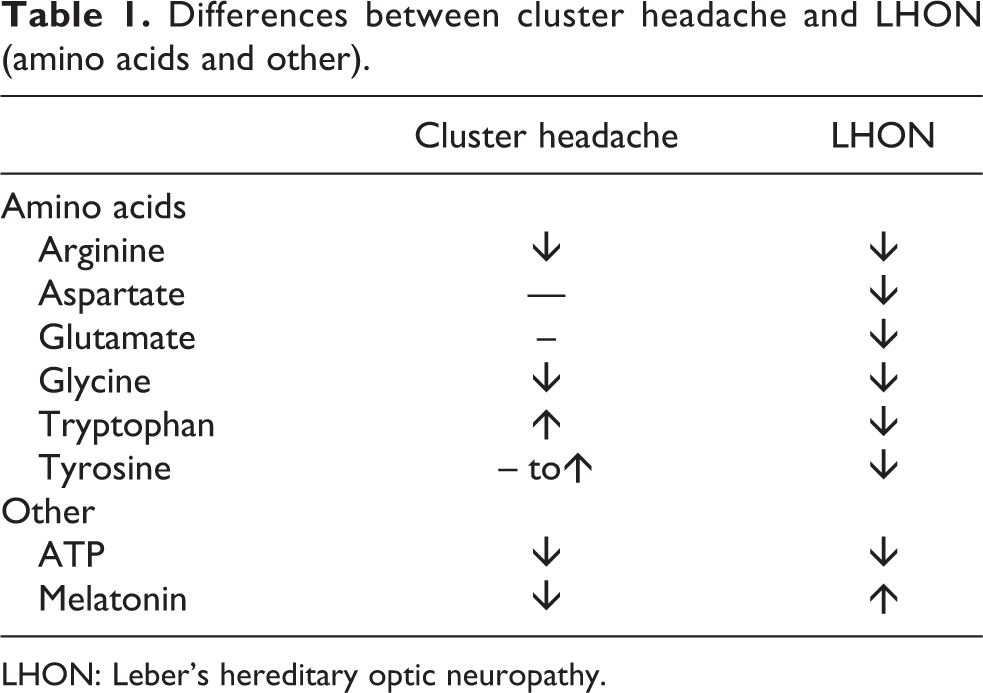
LHON: Leber’s hereditary optic neuropathy.
D’Andrea and colleagues have completed the most work on this subject noting that tyrosine, tryptophan, and arginine metabolism is abnormal in CH patients and this may be a metabolomics thumbprint for the disorder.
27
The possible mechanisms by which these metabolic alterations are involved in CH pathogenesis is found elsewhere, but they suggest mitochondrial dysfunction and energy failure in CH patients.
27
–29
The global decrease in amino acid levels in LHON patients may thus be protective against the development of CH as follows: Tyrosine: In CH patients, tyrosine levels are normal to elevated.
27,28
In addition, tyrosine metabolism is altered secondary to shifts in the activity of tyrosine decarboxylase and hydroxylase enzymes.
27
This results in changes in dopamine, norepinephrine, and elusive amine levels. All of these alterations may play some role in CH pathogenesis. Low tyrosine levels noted in LHON patients could potentially block these fluctuations in tyrosine metabolism and thus be protective against the development of CH.
27,28
Tryptophan: Tryptophan is the amino acid precursor of serotonin and its metabolites modulate N-methyl-D-aspartate (NMDA) receptors via the kynurenine pathways.
30
Tryptophan levels have been found to be significantly elevated in CH patients.
30
Serotonin levels are also significantly increased, while kynurenine levels are suppressed.
29,30
These findings suggest that there is an activation of the serotonin system along with a state of NMDA receptor hyperactivity in CH patients.
30
Recently, serotonin has also been shown to be a direct and indirect inhibitor of orexin neurons.
31
The hypothalamic neuropeptide orexin may play a significant role in CH pathogenesis and cerebrospinal fluid (CSF) orexin levels have been found to be reduced in CH patients, although the functional meaning of this finding has not yet been determined.
32
The suppressed tryptophan levels noted in LHON patients could thus be protective against the development of CH by altering serotonin and kynurenine pathways and possibly by modifying orexin secretion. Glutamate: Glutamate and its receptors may play a role in CH pathogenesis.
29,30
Orexin neurons are tonically activated by glutamate.
25,26,32
In addition, tobacco exposure elevates glutamate levels in smokers and nicotine stimulates orexin neurons.
33
The author has previously suggested that oxytocin may play a role in CH pathogenesis, possibly being involved in the activation and suppression of CH attacks.
4
Hypothalamic oxytocin release is triggered by glutamate and glutamate receptor antagonists blocks oxytocin release.
34
Thus, thus having low glutamate levels as a mutational consequence could be added protection in LHON patients from developing CH. Total amino acid suppression: The nonessential amino acids (in order of potency): glycine, aspartate, alanine, serine, asparagine, proline, and glutamine have all been shown to activate orexin neurons and promote locomotion in animal models.
35
All of these amino acids are decreased in LHON sufferers and thus another possible protective mechanism by which LHON patients do not develop CH.
Suggested treatment protocol for CH patients based on LHON mtDNA mutation consequences
It is feasible that if we can mimic some of the mutational consequences of LHON in CH patients, it could change the natural course of the disease. A proposed CH lifestyle regime is presented: Do not smoke especially if you had secondary smoke exposure as a child and/or stop smoking if you have already developed CH Maintain high melatonin levels at all times: The safety and tolerability of up to 30 mg per day of melatonin in TAC patients and 40 mg–100 mg per day in cancer patients has already been published.
36,37
One could consider starting all CH patients on melatonin and possibly targeting higher dosing strategies; however, this proposal will need further controlled trials to assess efficacy. Do not disrupt the retinohypothalamic tract: Many CH sufferers are alcohol users and abusers. Alcohol itself will acutely trigger a CH attack, especially in male smokers.
38
In animal models, it has been noted that acute alcohol use disrupts the phase resetting responses of the circadian clock to photic (glutamate) and nonphotic (serotonin) input, while chronic ETOH exposure significantly disrupts photic resetting of the circadian rhythm.
39,40
Alcohol seems to target the SCN directly as well as the retinohypothalamic tract. Abstinence from alcohol is suggested based on these findings as this may change the natural history of CH. Create a CH preventive diet: Is there a food diet that CH patients could follow which would help them match the metabolomic profile of LHON? ∘ Recently, the ketogenic diet/Atkins modified was noted to be effective in treatment refractory chronic CH in an open label study.
41
The authors hypothesized that the positive effect of the diet may have been secondary to an increase in dopamine and GABA levels induced by ketosis. Based on the LHON protective theory for CH, there may be an alternative explanation. The ketogenic diet enhances mitochondrial biogenesis and increases ATP production. In regard to amino acids changes on the ketogenic diet, a CSF study in epileptic children noted a decrease in tyrosine, glutamate, asparagine, serine, taurine, glycine, alanine, and phenylalanine levels, while a subsequent study noted that serum tryptophan levels were also lowered.
42,43
Thus, a global amino acid level suppression on the ketogenic diet mimics the metabolomic profile of patients with LHON. This will need to be evaluated further in more controlled studies. Enhance mitochondrial efficacy/Increase ATP production: Three available choices would include riboflavin (400 mg per day), coenzyme Q10 (200–1200 mg per day) and idebenone (900 mg per day). The latter two have already shown efficacy in migraine prevention but have never been extensively studied in CH. Idebenone is actually the only approved compound for LHON. It is a benzoquinone which can overcome mitochondrial complex I failures by transferring electrons directly to complex III of the electron transport chain and thus bypassing complex I altogether.
44
Idebenone treatment results in enhanced ATP production. In addition, it also resorbs reactive oxygen species.
44
It is unknown whether CH patients have specific complex I deficiencies and thus would be helped by idebenone treatment. Consider estrogen modulation therapy: LHON and CH are male predominant disorders, thus does estrogen create a protective effect from developing these conditions in women? Clomiphene citrate, a hypothalamic estrogen receptor modulator, has shown possible efficacy as a preventive treatment for refractory CH.
45,46
Estrogen treatment has shown effectiveness in laboratory models of LHON (enhancing mitochondrial biogenesis and activating superoxide dismutase) and more recently in patients.
47
Finally, melatonin has demonstrated antiestrogenic effects via ERα pathways.
48
Thus adding in an estrogen modulatory treatment to CH patients may alter the natural history of the disease.
All of these treatment suggestions currently lack strong scientific data and thus are options alone and not a specific protocol that should be utilized in the clinic until further study is completed.
Conclusion
Looking beyond headache patients to other neurologic conditions may end up leading to a better understanding of headache pathogenesis. Patients with LHON have a higher than average chance of developing CH, but there are no reported cases in the literature. Possibly, there is no cause and effect relationship or maybe the consequences of the mtDNA mutations noted in LHON are actually protective against the development of CH, especially in smokers. This information would truly expand on our current knowledge of CH pathogenesis and possibly provide new therapy options for a very disabling headache condition.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.