
Abstract
In this news and views, we discuss our recent publication where we described how ER-PM membrane contact sites (MCS) are modulated during store operated calcium entry (SOCE). We also examine why enforcing ER-PM MCS by tethering proteins does not not enhance, but rather inhibits SOCE.
Keywords
Membrane contact sites (MCS) forming between the endoplasmic reticulum (ER) and the plasma membrane (PM) or intracellular organelles drive fundamental physiological processes by sustaining calcium signalling and enabling lipid transport between membranes. MCS are sites of close apposition (10–30 nm) of two membranes joined by protein tethers, serving as platform for the non-vesicular transfer of lipids and for molecular interactions between ER and PM proteins that control the ubiquitous store-operated Ca2+ entry (SOCE) cell signalling pathway (Balla et al., 2020). The dynamic changes in membrane lipids and local Ca2+ concentration at MCS are finely tuned in time and space by the activity of transporters and their regulatory proteins, enabling these elemental cellular structures to regulate a wide array of cellular functions.
MCS are dynamic structures that form, elongate, retract, and detach in response to cellular cues by the coordinated actions of tethering and signalling proteins. This poses a challenge when studying MCS structure-function relationship, as the ultrastructural alterations occurring during MCS biogenesis need to be precisely resolved and quantitatively related to the cellular functions driven by MCS-supported processes. Resolving MCS morphological changes is difficult with optical approaches, and numerous studies report alterations in MCS structures of little functional significance and functional defects occurring without changes in MCS structure. In a recent study, we attempted to fill this gap in knowledge by performing a quantitative and systematic evaluation of the ultrastructural changes occurring at MCS during the SOCE process using the gold standard of electron microscopy (Henry et al., 2022).
SOCE is triggered physiologically following agonist stimulation by the generation of inositol trisphosphate that releases Ca2+ from the ER into the cytoplasm. The ER Ca2+ depletion causes a conformational change in the ER membrane-resident proteins stromal interaction molecules 1 and 2 (STIM1 and 2), releasing an intramolecular clamp to promote protein extension, oligomerization, and the exposure of a polybasic tail with lipid affinity. The activation switch enables STIM proteins to translocate to ER-PM MCS, where they trap and gate the Orai family of Ca2+-selective channels. Orai channel activity floods the cytosol with Ca2+ ions and enables the refilling of ER calcium stores by sarco/ER Ca2+-ATPase (SERCA) pumps near the MCS, thereby terminating SOCE (reviewed in Qiu and Lewis (2019))
The formation of macromolecular clusters containing STIM-bound Orai channels occurs at ER-PM junctions with gap distances ranging from 10 to 30 nm. Shorter gaps generated by chemically inducible linkers prevent the access of the large Orai1 channel into MCS (Varnai et al., 2007). The gap distance is dynamically regulated by the Ca2+-dependent tethering protein extended synaptotagmin 1 (E-Syt1), which when overexpressed in COS-7 cells accumulates in ER-PM junctions during SOCE and mediates a ∼8 nm reduction in gap distance (Fernandez-Busnadiego et al., 2015; Giordano et al., 2013). In HeLa cells SOCE induction increases the density of cortical ER (cER) structures 5-fold and promotes the appearance of characteristic long and thin cER sheets enriched in STIM1 while in MEF cells SOCE increases the cER length 2-fold when STIM1 is overexpressed (Orci et al., 2009; Sauc et al., 2015). The functional significance of these ultrastructural rearrangements is unclear as it is difficult to predict the impact of gap shortening and of cER elongation and narrowing on local and global Ca2+ fluxes. For instance, at the ER-mitochondria interface, enforcing massive tethering induces cell death and reducing the gap distance below 10 nm decreases ionic fluxes by preventing the access of ion channels to the narrowed MCS (Csordas et al., 2010).
To assess the functional significance of the cER rearrangements occurring during SOCE we quantified by electron microscopy the length and gap distance of individual contact sites forming during exposure of cells to the SERCA ATPase inhibitor thapsigargin. We imaged cells at three phases of this standard SOCE protocol: In physiological saline before ER depletion (NT), 10 min after ER depletion in a Ca2+-free solution (CF) and two minutes after Ca2+ re-addition (CA), to separate the effect of ER depletion and of calcium entry (Figure 1A). This strategy revealed that the gap distance remains stable throughout the SOCE protocol while a fourfold increase in ER-PM length was observed following ER depletion that persisted after Ca2+ readmission (Figure 1A). We then attempted to promote the formation of contacts by expressing two different E-Syt isoforms shown to either promote (E-Syt1) or not (E-Syt2) a Ca2+-dependent gap shortening (Giordano et al., 2013). As alternative strategy we used artificial ER-PM tether proteins generated by the group of Jen Liou, based on the TM domain of STIM1 for ER retention and on the polybasic motif of the small G protein Rit for constitutive binding to PM phosphoinositides (Chang et al., 2013). We used two versions of these synthetic tethers, a long version (MAPPER-L) bearing flexible cytosolic linkers meant to preserve the endogenous 10–25 nm ER-PM gap distance of STIM-Orai1 contact sites and a shorter version (MAPPER-S) designed to accumulate in junctions of 10 nm. Electron micrographs of cells overexpressing either endogenous or artificial tethers revealed that the gap distance was only marginally reduced during ER Ca2+ depletion while the cER expansion associated with the activation of the SOCE pathway was greatly potentiated by the tethering proteins, an effect that was partially reversible upon Ca2+ readmission (Figure 1B and C). Greatly elongated (>300 nm) cER sheets were observed in cells expressing tethering proteins that were anchored at tether-specific distances of 12–15 nm (E-Syts) and 5–9 nm (MAPPERs). The MAPPERs are therefore not passive cER markers as generally assumed but facilitate the expansion of the cER during SOCE by anchoring cortical ER sheets at specific gap distances from the PM. Of note, store-operated cER expansion was observed in cells selected for low MAPPERs expression levels by flow cytometry. Artificial junctional structures can be observed at high MAPPERs expression levels (Changet al., 2017 and our unpublished observations), suggesting that the impact of the constitutive tethering is dose-dependent.

Changes in Cortical ER Length and gap Distance During Store-Operated Ca2+ Entry. A. Changes in Cytosolic Ca2+ Concentration (Black Trace), cER Length (Yellow Trace) and gap Distance (red Trace) During the Depletion of ER Ca2+ Stores with Thapsigargin (Tg) in Calcium-Free medium (CF) and the Subsequent Readmission of Ca2+ to Evoke SOCE (CA)
Unexpectedly, the expansion of the cortical ER enabled by the tethering proteins did not enhance, but inhibited SOCE. The rates of store-operated Ca2+ entry, measured with fura-2 using either Ca2+ or Mn2+, increased as the cortical ER elongated in wild-type HEK-293 cells but not in cells expressing the MAPPER constructs (Figure 1D). A key to this conundrum was given by the respective locations of the tethering and signalling proteins. The two MAPPERs assembled in clusters contiguous to STIM1 and Orai1 clusters forming in the TIRF plane, while the non-fusogenic SNARE protein Sec22b, whose expression does not impact SOCE, co-localized extensively with STIM-Orai1 complexes. These findings indicate that STIM1/Orai1 complexes do not populate cER sheets anchored at gap distances shorter than 10 nm from the PM by protein tethers (Henry et al., 2022), consistent with earlier findings using rapamycin-induced gap shortening (Varnai et al., 2007). The lateral extension of the tethered cortical ER sheets excluding STIM-Orai1 complexes then appears to have an inhibitory effect on SOCE.
In summary, by resolving the ultrastructural changes occurring at ER-PM contacts sites during SOCE, we showed in our recent study that the cER elongates as endogenous STIM and Orai proteins interact at MCS and that this cER extension persists during Ca2+ elevations resulting from Ca2+ entry. The cytosolic Ca2+ elevation still had a regulatory role as it slightly decreased the length of the extended contact generated in the presence co-expressed the tethers. Although these structures might enable the transport of lipids between the two membrane, they do not allow the formation of STIM-Orai1 complexes, likely because they are too narrow to accommodate the large cytosolic domain of Orai channels (Figure 2). One limitation of our study is that overexpression of tethers might decrease SOCE due to a displacement of the endogenous machinery. Alternatively, the overexpressed tethers might displace the endogenous complexes by saturating STIM1 lipid binding sites on the PM. Another limitation is that these morphological changes were observed with the SERCA inhibitor thapsigargin, which we used to evoke a prototypical SOCE response. Further studies should attempt to document the ultrastructural changes in cER occurring during physiological stimulation of cells with agonists.. Another point to be raised is that these morphological changes were observed with the SERCA inhibitor thapsigargin, which we used to evoke a prototypical SOCE response. Further studies should attempt to document the ultrastructural changes in cER occurring during physiological stimulation of cells with agonists.
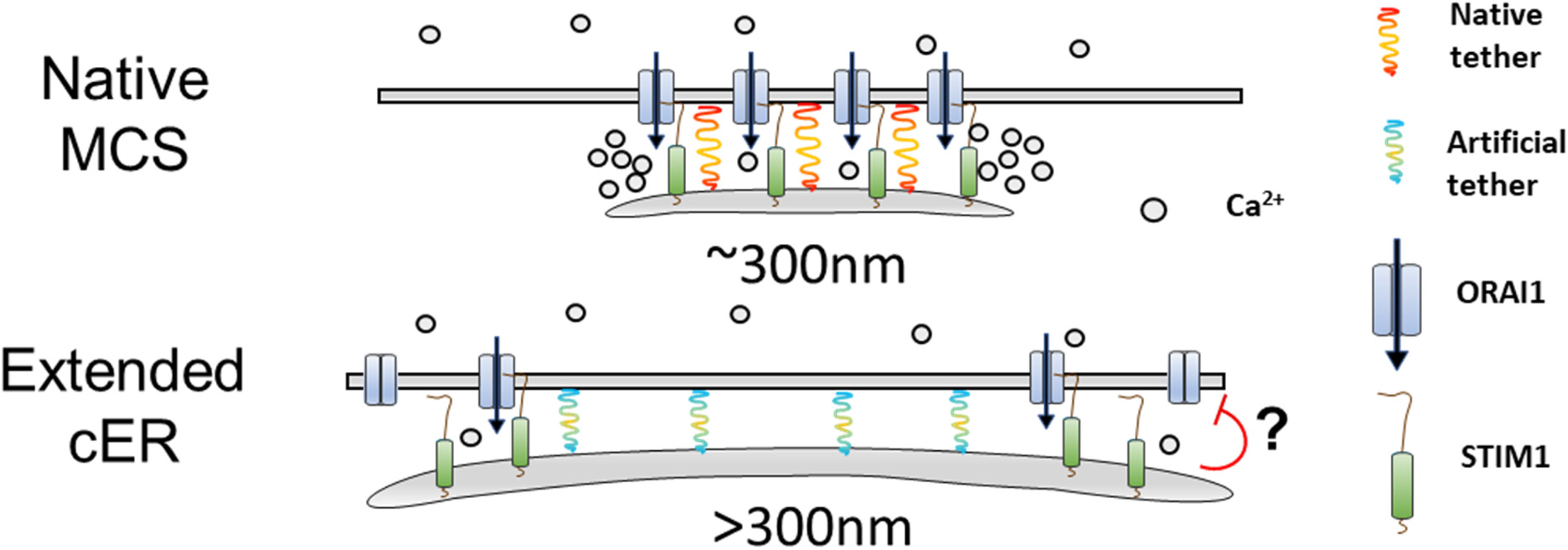
Enforced cER extension inhibits store-operated Ca2+ entry. Top: cER sheets elongate during store depletion to a maximal length of 300 nm and remain 18 nm away from the PM during Ca2+ elevations, a distance sufficient for STIM1-Orai1 complexes to form along the whole ER-PM interface. Bottom: Artificial or overexpressed tethering proteins promote the expansion of cER sheets anchored <10 nm from the PM, confining STIM1-Orai1 complexes to the rims of cER sheets at locations that might promote the Ca2+-dependent inactivation of Orai1 channels by accessory proteins. Modified from (Henry et al., 2022).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (grant number 310030_189042 (to ND), Novartis (22B082), Stiftung für die Erforschung der Muskelkrankheiten, Sir Jules Thorn Charitable Trust, , (to A C-S).