
Abstract
Background
Safe and effective biosimilar medications have the potential to significantly increase access to these valuable drugs. The two current biosimilars available in dermatology in the United States (US) are infliximab and rituximab which were Food and Drug Administration (FDA) approved in 2016 and 2018 respectively. There has been significant interest in this topic as a number of biosimilar versions of adalimumab will be available in 2023.
Objective
This review will discuss biosimilar basics and the experience with biosimilars used in dermatology in the US, Asia, and Europe.
Methods
All articles in Ovid/Medline from 2015 to Feb 2023 on biosimilars were reviewed with a particular emphasis on medications used in dermatology. Other reports from pharmaceutical manufacturers and blogs following the development of the biosimilar industry provided key insights.
Results
Biosimilars have been able to produce significant savings and market share increases, particularly in Europe, where there has been a longer experience. The specifics depend on drug prescribing practices and incentives in the individual country. This degree of savings and market share increases have not been realized with the current biosimilars available in the US.
Conclusion
While biosimilars have resulted in significant savings compared to originator drugs, it is clear that prescribing incentives and physician education are crucial in achieving these savings. To what degree biosimilar market share will increase in the US remains to be determined.
Introduction
The term biosimilar was coined by the European Medicines Agency because the drugs had similar efficacy and safety but were slightly different. A biosimilar is a biological product that is highly similar to, and has no known clinically meaningful differences from, an existing FDA approved reference product. It is not the same as a generic drug as these are more complex molecules manufactured via living organisms. In the lifecycle of manufacture, biologic medications undergo slight changes due to post translational modification so the products are not identical.
A simple analogy is a lock and key. A biosimilar product will unlock the same key as the reference product but the other end of the key may have slight differences. These slight differences are known as the microheterogeneity. There have been concerns raised over microheterogeneity as these products can change slightly over time due to factors such as varying cell lines, manufacturing techniques, and more. 1 These changes could lead to decreased efficacy via immunogenicity or increased safety concerns. 2 These slight post translational modifications over time have prompted FDA to continue monitoring the safety and efficacy of both originator and biosimilar products. It is known that the brand name adalimumab (Humira) that was prescribed in 2010 is not identical to the same brand name product that was recently prescribed due to this same issue. 3 As such this concern is not unique to biosimilar medications and is instead a matter of pharmacovigilance for all biologic drugs, originator and biosimilar.
The FDA gained authority to approve biosimilars in 2010, when the Biologics Price Competition and Innovation Act (BPCIA) entered into law, creating an abbreviated approval pathway for biosimilars. Since the passing of the BPCI Act, multiple biosimilars have gained FDA approval and entered the market. All biosimilars will have a four-letter suffix such as atto or adbm in the case of adalimumab and axxq and dyyb in the case of infliximab
After a biosimilar product is shown to demonstrate the desired target binding (unlock the key) then significant studies follow to show safety, purity and potency. These are conducted to measure immunogenicity, pharmacokinetics, pharmacodynamics, and subsequently safety and efficacy through clinical studies. If the biosimilar is approved then the manufacturer needs a specific pharmacovigilance plan to continue to monitor these attributes. 4
When biosimilars undergo testing to compare to the originator product, they are studied in the context of clinical equivalence margins. This means that the equivalence of the biosimilar to the originator is compared within a predetermined range. This range is determined using a variety of both clinical and statistical criteria that are beyond the scope of this review. In determining these margins, comparisons of the originator to placebo can be helpful as the margin should always be smaller when biosimilars are compared to the originator. In the absence of ample prior data to determine a margin, regulators have suggested an equivalence range of 80-125%. 5
It is important to understand the terms extrapolation and interchangeability with respect to biosimilars. When a biosimilar is approved for one disease state it may also be approved for other disease states the originator product is also approved for. This is determined by the responsible regulatory agency. As an example, when a biosimilar version of infliximab was granted approval for rheumatoid arthritis based on all the preliminary investigations and clinical studies, the FDA also granted approval in Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis.6-9 Interchangeability refers to the ability to switch to a biosimilar from its reference biologic without prescriber authorization. In general, additional studies are required with the biosimilar product to be granted this interchangeability label. Interchangeability rules vary significantly based on the country and state. Many states in the US do not allow substitution without prescriber authorization even with the interchangeability label.10-12
There has been initial physician apprehension regarding the use of biosimilars. These include concerns of the quality of biosimilar medication, the safety data base being insufficient at the time of licensing, increased immunogenicity, the efficacy being different from the reference product and switching from reference to biosimilar medication.13,14
Alongside hesitant physicians, manufacturers of originator drugs have raised several potential limitations with biosimilar studies. The studies with biosimilars are smaller and underpowered compared to the pivotal phase III studies for originators and therefore may miss rare adverse reactions. The bioequivalence may be under powered based on the small sample size. There have also been arguments against extrapolating data for treating one disease in order to approve treating another. If the biosimilar study is done in rheumatoid arthritis will the same biosimilar work as well in psoriasis or Crohn’s disease? Is drug retention in biosimilar studies a suitable surrogate for safety and effectiveness? The concern has also been raised over selection bias in some studies as copay assistance could be available for the originator, but not for the new biosimilar. 15 Ultimately these concerns have not led to documented inferiorities of biosimilars. There have been >90 switching studies performed with biosimilars which have not revealed any significant issues.13,16-33
This continued surveillance must remain a focus of pharmacovigilance programs to monitor biosimilars as they are on the market. 34
Europe
In 2005 the European Medicines Agency created the framework for a biosimilar approval process. This early adoption led to the European Medicines Agency approving over a dozen biosimilars before the USA had its first biosimilar approval in 2015. 35 The first biosimilar approval in Europe was in 2006. The first monoclonal antibody biosimilar approval was infliximab in 2013. 36 Having one approval process that spans all countries in the European Union allowed for widespread adoption across the continent, but there are still considerable differences in biosimilar uptake depending on the country. This is largely due to the European Medicines Agency providing marketing authorizations for biosimilars, but leaving further regulation to individual countries. One major regulation left to countries is approving interchangeable status of a biosimilar with its originator product. Other regulations that can affect biosimilar uptake include tendering processes, provider incentives, and prescribing quotas for biosimilars. 37
Europe has led the way to introducing and increasing access to biosimilar medications. There has been a 14-year experience in Europe with biosimilars. There are 70 biosimilars approved. There has been over a 2 billion patient day exposure to biosimilars there.11,38,39 The uptake varies significantly between countries with over 90% biosimilar adalimumab and infliximab use within in 1-2 years in some of the Scandinavian countries and 40 to 50% over many years in countries such as France, Italy and the UK. 40 There was a more rapid uptake of etanercept in Europe which was launched a few years after infliximab likely because of increasing physician comfort with biosimilar use. Biosimilars of rituximab have achieved a 50% market share in top EU markets after less than 2 years. 41 In terms of specific countries within a year of launch, the United Kingdom and Netherlands have more than 90% penetration by rituximab biosimilars. 42 For most European Nations, biosimilar use now far outnumbers originator brand prescriptions each year. 42
Price decreases with biosimilars in Europe have varied but is some cases cost savings up to 80-90% have been demonstrated. These aggressive savings are seen in single payer systems who have strong positions of price negotiation due to their size and uniformity. Whether these types of savings can be achieved in the US is unlikely.
A potential major advantage of biosimilar availability is increased access which hopefully leads to better patient outcomes. 36 As examples in the United Kingdom G-CSF use increased by 228% and in Bulgaria the use of the anti-TNF agents etanercept and infliximab increased by 163%. 43
Asia
Multiple studies have tracked measures of biosimilar use and acceptance across Asian countries. Like Europe, the acceptance of biosimilars appears to be related to the cultural and institutional encouragement of their use. South Korea has emerged a world leader in biosimilar development. 44 Over the past decade the government has provided significant capital and regulatory assistance to biosimilar companies in order to facilitate biosimilar development. 44 One study, which surveyed physicians across eight Asian countries, found that a majority of these physicians understood that biosimilars are similar to the originator, but not equivalent. However, over a third surveyed also felt that immunogenicity was a negative aspect of biosimilars compared to originators. 39% of physicians from Asian countries surveyed responded that they were “not confident” or “a little confident” in using biosimilars in clinical practice. There were also significant regional differences in the perceived financial benefits. Nearly half of Korean physicians surveyed believed biosimilars would only have a marginal impact on drug prices, compared to just 5.9% in Chinese physicians. 45 Like Europe, the uptake of biosimilars appears to have significant regional differences, but unlike European countries, there is not a central medical agency in Asia which can approve a biosimilar for multiple countries at once.
United States
There are currently four FDA approved dermatology biosimilars in the US, infliximab, etanercept, adalimumab, and rituximab. Only biosimilar infliximab and rituximab have been used in clinical practice, infliximab since 2016 and rituximab since 2018. There are two biosimilars of the originator, etanercept, that are FDA approved but waiting to launch. Eticovo™ created by Samsung Bioepis currently has no known launch date as it is still in litigation. Attempts to launch the product were set back after Sandoz, the other company with an etanercept biosimilar awaiting launch, Erelzi™, failed to prove that two patents held by Immunex related to etanercept were invalid. While the launch date of Eticovo™ is not yet determined, a federal court has ruled that Erelzi™ made by Sandoz will not launch until 2029.
Adalimumab Biosimilars Release Time Frame.
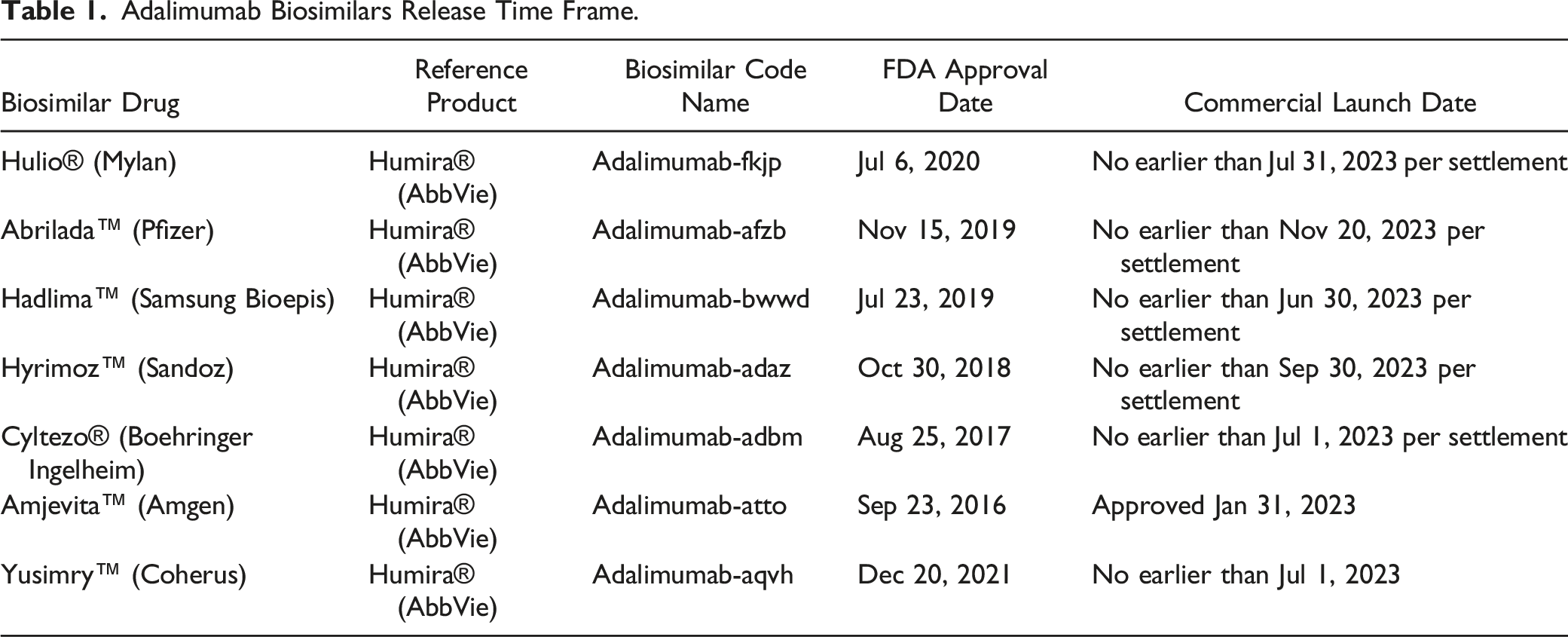
Further, there has been progress made on several ustekinumab biosimilars.47-49 The patents for ustekinumab will be expiring in 2023 in the USA, and 2024 in Europe, allowing for another class of biosimilars to enter the market. 50 While there are currently no FDA approved biosimilars of ustekinumab, both Alvotech and Amgen have biosimilar products under review by the FDA, along with another 6 biosimilars in development. 51
Changes and Updates in Laws
Most recent updates in the laws regarding biosimilars come as the US regulators and market attempt to further streamline the entry of biosimilars to the market. A large step in this came from the release of the joint statement by the FDA and FDC on Collaboration to Advance Competition in the Biologic Marketplace which was released February 2020.
These guidelines included materials (1) to promote greater competition in biologics markets, (2) to deter behavior that impedes access to samples needed for biologic development, (3) to take appropriate action against false or misleading communications about biologics, and (4) to review patent settlement agreements involving biologics for antitrust violations. These new guidelines come as a multitude of lawsuits have been used to determine future release dates for new biosimilars. With these specific guidelines, there may be less litigation in future biosimilar rollouts. 52
The FDA also released guidance on Promotional Labeling and Advertising to prevent false claims regarding biosimilars. This came from lawsuits that stemmed from certain wording on biologic products that could be construed to imply that they perform differently than biosimilars of the same originator, which is explicitly false given that the current approval system for biosimilars requires the biosimilar to be proven clinically equivalent to the originator.
In February 2020, FDA issued draft guidance titled “Biosimilars and Interchangeable Biosimilars: Licensure for Fewer Than All Conditions of Use for Which the Reference Product Has Been Licensed.” This states that while a biosimilar cannot be approved for uses other than those approved for the reference drug, it can be approved for less than the reference drug. This allows biosimilar manufacturers to save time and money on testing biosimilars as they can focus on testing the medication only for conditions that it will be approved to treat.
Current Dermatology Biosimilar Use in United States
While oncology biologics and supportive biologics such as filgrastim and epoetin alpha have had biosimilars taking between 25-50% of market share, biosimilars that prove useful for dermatologic treatments have had a slower entry into the market. 52 This has largely been attributed to complex interactions between pharmaceutical manufacturers, insurers, and pharmacy benefit managers. 15 Due to the fragmented nature of medical insurance in the USA, it is difficult to achieve the bulk savings seen with biosimilars in other countries. 53
Until 2023, the only two biosimilars available in dermatology were infliximab (for psoriasis and psoriatic arthritis) and rituximab (for pemphigus). The refence products were approved in 1998 and 1997 respectively and the biosimilars in 2016 and 2018. As of 2020, infliximab biosimilars have only achieved 20% of market share by volume despite being available since 2016, earlier than many other biosimilars in the USA. 52
In our initial experience with biosimilar rituximab and infliximab there was very little pressure to switch because of the minimal cost differential. In the last 2 years certain payors have mandated the biosimilar product for new starts. In some of these cases the payor mandated a higher patient payment/copayment for the patient to be started on the reference product (financial incentives directly to the patient to start with a biosimilar). Recently there have been requests to switch a number of our patients on the originator drugs to the biosimilar version. In New York State this substitution cannot be made without physician authorization. As mentioned above, the interchangeability guidelines vary significantly by country and state. Our anecdotal experience with switching from the originator to a biosimilar has not demonstrated any significant issues with side effects or loss of efficacy.
Barriers to Increase Biosimilar Use in United States of America
A persistent challenge in biosimilar uptake has been physician awareness and knowledge of biosimilars. A recent survey of American physicians revealed that fewer than half of physicians understood the core concepts of biosimilars, even among physicians who have previously prescribed them. Also half of physicians surveyed indicated they were hesitant to prescribe biosimilars until they had been on the market longer. 28 Insurance formularies create a complex barrier accessing biosimilars. While it is easiest for healthcare providers to purchase bulk quantities of one type of drug, insurance formularies demand the opposite situation, one in which patients are required to have access to different versions of drugs of the same family. This creates a situation in which a pharmacy benefit manager may have to stock a variety of versions of the same drug in order to satisfy the array of insurance plans presented by patients. This problem reduces the cost saving effects of biosimilars and competition between drug manufacturers. This also can create a frustrating situation for prescribing physicians, who may shy away from new biosimilars wanting to avoid more complexity and confusion within a given plan.
The storage requirements for biologics also cannot be ignored as many have individual storage requirements. By doubling or tripling the number of biologics that need to be on hand to satisfy payer formularies, the cost of storing these drugs could diminish any cost savings from using biosimilars.
Frequent switching of insurance formularies also increases the need for scrutiny at each dispensing of medication, consuming more time to deliver drugs to patients. Additionally, changes in formularies, or patients changing insurance carriers, might result in a healthcare plan no longer having medications covered available, resulting in delays in patient treatment. This is especially problematic in oncologic therapy, which is heavily dependent on biologics, when prompt treatment can be crucial in treatment efficacy.
Attempts to Increase Biosimilar Use in United States of America
It is hoped that the introduction of biosimilars in dermatology will increase access of these valuable medications and decrease the overall cost to the healthcare system. This statement is from the Task Force on the Use of Biosimilars to Treat Rheumatological Diseases “The availability of biosimilars must significantly lower the cost of treating individual patient and increase access to optimal therapy for all patients”. 54 To what degree this can be achieved in the United States remains to be determined.
Clearly physician education regarding the safety and efficacy of biosimilars is crucial. The most agreed upon actions for increasing biosimilar use in the United States were increased education of providers, streamlining the biosimilar prior authorization process and incentivizing their use. 13
In Europe there are individual countries with over 80% biosimilar adalimumab use and over 50% cost savings. The primary driver for this increased use is government incentives and price reduction through competition leading to increased utilization due to the lower price. 55 Hopefully with the number of biosimilar versions of adalimumab expected in 2023 this type of competition and price reduction will occur. The incentives will likely come from payors and pharmacy benefit managers. Practice settings and hospital ownership status currently had the largest associations with biosimilar usage in the United States. 56
At present there are 11 injectable biologic agents available for psoriasis in the United States. Initially, the agents in the biosimilar category for dermatology are the anti-TNF agents infliximab and adalimumab. Biosimilar etanercept is not expected until 2029. Biosimilar rituximab is approved for pemphigus but is primarily used in oncology. The availability of biosimilar infliximab since 2016 has not been a significant issue since the overall use of this drug is very low in dermatology. The uptick in infliximab use in rheumatology and gastroenterology has also been slower than in most European countries. The use of adalimumab, the number one selling drug in the United States, will definitely be a different situation as the biosimilar versions come to market considering the high utilization and the multiple indications approved for this drug. One conservative estimate of biosimilar adalimumab use in the United States estimated over 2 billion dollars in Medicare savings over a 4 year period. 57
As in our experience with initial approval of biosimilar agents there may be little price differential and little incentive for use. Even with a price difference, with rebates and company incentives there are less out of pocket costs for the patient to remain on the brand name product. It is also not clear what price difference (10%, 30%, 50%) will be the magic number to create the most incentives. Any rebates and discounts of biosimilars should be shared with patients. Patients will not appreciate if an originator is switched to a biosimilar for cost savings that they will never see.
The difficulty with step edits and prior authorization is already a significant burden on our practices. How this will change with the introduction of biosimilars is also an important issue. Our experience is that currently new starts for infliximab and rituximab are mandated to be biosimilar but these biosimilar products have already been approved for use for over 5 years. Initially there was little or no pressure to start or switch. How quickly will biosimilar adalimumab be part of the step edit process will depend on Medicare or the individual payors involved but will be another key driver. As a specific example we recently received this notification from an insurance company’s prior authorization approval for brand name Humira “If your approval is for a brand/biologic agent your authorization may be approved for a generic biosimilar if one becomes available on the market before your authorization expires”.
Another important issue to be determined is the formulary status of biosimilars with the different payers and pharmacy benefit managers. Within the 11 injectable biologic products available for psoriasis, there are essentially three classes, the anti-TNF agents, the anti-IL 17 agents, and the anti-IL 23 agents. In some cases, all eleven of these drugs are on a given formulary. In many cases there are restrictions, such as one drug within each class preferred. Many plans have opted to incentivize one or two agents within a class of drugs based on the manufacturer rebates they receive and the percentage of use within the class.
It is likely that at some point biosimilar adalimumab will be the preferred agent within the anti TNF class for many payors. Another possibility could be that biosimilar adalimumab will be a mandatory step edit before a patient can receive an anti-IL17 or anti-IL23 drug despite the fact that the treating physician feels there are specific advantages with one of these other agents based on the patient’s history, efficacy and side effect issues. 15
Unfortunately, we are already dealing with this type of issue as some plans continue to mandate previous methotrexate or phototherapy use before a biologic agent is approved. Our hope is that the physician prescribing decisions about the treatment of choice for an individual patient are honored. For a patient with psoriasis or psoriatic arthritis for whom adalimumab is the drug of choice the introduction of biosimilar adalimumab will likely increase access and decrease costs. To what degree these goals can be achieved will be determined over the next few years.
Conclusions
While biosimilars have been increasing in popularity and market share in the USA, there is still much more room for growth, and a litany of biosimilars awaiting commercial launch or FDA approval. Recent actions taken by the FDA and FDC pave the way for more streamlined biosimilar launches which could significantly lower the cost of biologic drugs in the US. Physician education and potential cost savings will play a key role in the continued increase in the use of biosimilar medications.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Robert E Kalb has received research funding from AbbVie, Amgen, CoreEvitas, LLC, Janssen-Ortho Inc, University of Pennsylvania, and UCB. He has been a consultant for Janssen-Ortho Inc. and UCB. He has been on the Dermatology Safety Monitoring Committees for Lilly and PPD.