
Abstract
Introduction
Niemann-Pick disease is an autosomal recessive lysosomal storage disorder secondary to a deficiency of acid sphingomyelinase (ASM).1,2 Five main subtypes of Niemann-Pick disease are recognized, with varying degrees of ocular involvement.1,2 A complete loss of function or a partial loss of function in the sphingomyelin phosphodiesterase 1 gene is implicated in Niemann-Pick disease type A and type B, respectively. Type A is usually fatal by 3 years of age because of the involvement of the central nervous system (CNS) and viscera. Type B is compatible with life and tends to affect only the viscera.1,2 Mutations in the Niemann-Pick C1 and C2 genes (NPC1 and NPC2, respectively) result in progressive neurodegeneration with developmental regression and are found in Niemann-Pick disease type C and type D, with the Nova Scotian population being affected by type D.1,2 Niemann-Pick disease type E has been proposed as an adult-onset subset of the condition.1,2
Ocular findings in Niemann-Pick disease vary based on the subtype, with the most common being macular halos, which were first described in 1982 by Cogan et al 3 as bilateral symmetric parafoveal granular deposits. Corneal opacification and lens capsule discoloration can also occur in Niemann-Pick disease type A. 4 Individuals with Niemann-Pick disease type C may exhibit supranuclear vertical palsy, optic disc pallor, and parafoveal discoloration that appears gray. 1
We describe a patient with Niemann-Pick disease type B with bilateral macular halos and the successful surgical closure of a concurrent full-thickness macular hole (FTMH). Cytopathology of the peeled internal limiting membrane (ILM) and vitreous provided additional data on the etiology of the macular halo.
Case Report
A 72-year-old emmetropic White man with no significant ocular history was referred for a 1-day history of reduced vision in the right eye secondary to an FTMH. The patient’s medical history included a chronic dry cough, dyslipidemia, and hypertension, for which atorvastatin and perindopril were taken regularly, and a splenectomy after a traumatic rugby accident. The patient’s family history did not include metabolic disease, and both of his adult children were healthy.
On examination, the patient’s visual acuity (VA) was 20/120 OD and 20/16 OS. The intraocular pressures were normal. An examination of the anterior segment was unremarkable. A dilated fundus examination with a retinal imaging system (Optos, Optos Inc) showed a pale-yellow halo of granular deposits encircling the fovea with a symmetric appearance in both eyes (Figure 1A). An MH was seen in the right eye. Fundus autofluorescence (Optos) was unremarkable other than a small hyperautofluorescent dot at the right fovea (Figure 1B). The halos appeared bright on red-free imaging (Visucam 500, Carl Zeiss Meditec AG) (Figure 1C) but were hyporeflective on near-infrared imaging (Spectralis HRA+OCT, Heidelberg Engineering GmbH) (Figure 1D).
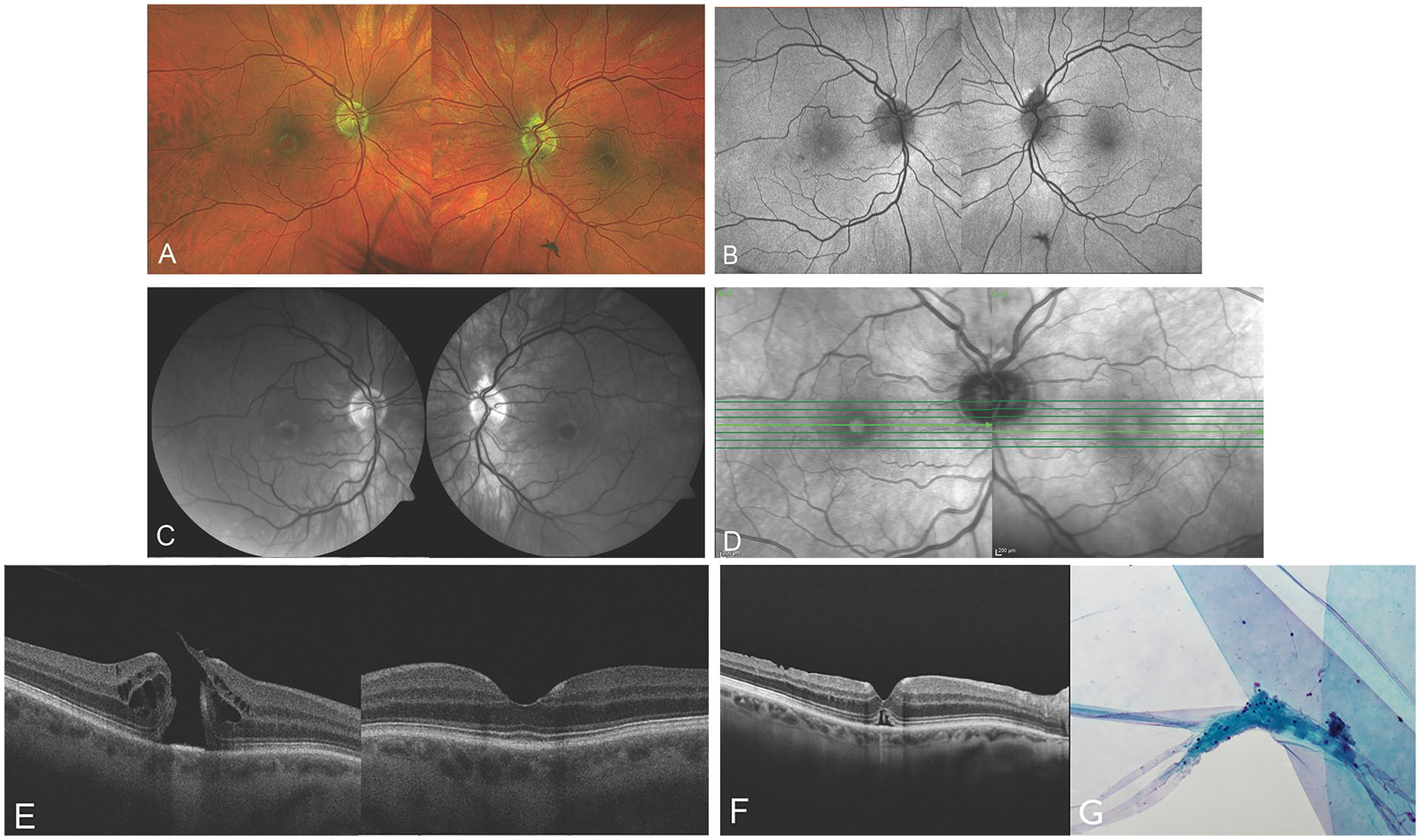
Multimodal imaging of a 72-year-old White patient. (A) Color fundus photograph shows bilateral macular halos and a macular hole (MH) in the right eye. (B) Fundus autofluorescence shows a small hyperautofluorescent dot at the right fovea but is normal in the area of the macular halos. (C) The halos appear bright on red-free imaging. (D) On near-infrared imaging, the halos are hyporeflective. (E) Spectral-domain optical coherence tomography shows bilateral focal inner retinal hyperreflectivity at the foveal slope and a full-thickness MH in the right eye. The foveal floor in the left eye is unaffected. (F) Six weeks after vitrectomy and inner limiting membrane (ILM) peeling, there is successful closure of the MH. The macular halo remains unchanged. (G) Cytopathology of the ILM (original magnification ×200) shows low cellularity and an absence of lipid deposits, suggesting the absence of inflammatory changes at the vitreoretinal interface. A tiny fragment of neural tissue is attached, as is often seen with ILM specimens.
Spectral-domain optical coherence tomography (OCT) (Cirrus HD-OCT 5000/500, Carl Zeiss Meditec AG) showed focal parafoveal inner retinal hyperreflectivity located at the foveal slope in both eyes that did not extend across the foveal floor in the left eye (Figure 1E). In the right eye, OCT showed a large (401 mm diameter) FTMH with vitreous attached to the margin of the hole. The average combined thicknesses of the ganglion cell layer (GCL) and inner plexiform layer (IPL) were 81 µm in the right eye and 79 µm in the left eye. High-resolution computed tomography of the patient’s chest showed interstitial pulmonary fibrosis, bibasal subpleural reticulation, and ground-glass opacification. Magnetic resonance imaging of the brain showed mild age-related, small-vessel, white-matter disease. Genetic testing identified a pathogenic variant in the sphingomyelin phosphodiesterase 1 (SMPD1) gene c.757>C (p.Asp253His) with heterozygosity, consistent with Niemann-Pick disease type B.
A vitrectomy with elevation of the posterior hyaloid, ILM peeling, and 26% sulfur hexafluoride gas tamponade were performed in the right eye, followed by 3 days of prone positioning. By 6 weeks postoperatively, the FTMH had closed, and the VA in the right eye had improved to 20/60 (pinhole 20/40) (Figure 1F). However, the macular halo and parafoveal inner retinal hyperreflectivity remained on OCT. Cytology (ThinPrep, Hologic) showed the peeled ILM had no specific features. A tiny fragment of inner neural retina was attached to the ILM, which is not unusual in ILM peeling specimens. The vitreous washings were low in cellularity, with a few hyalocytes and no other cells (Figure 1G).
Conclusions
The diagnosis of Niemann-Pick disease type B is often delayed, and individuals usually survive until adulthood. Systemic involvement includes splenomegaly, dyslipidemia, and pulmonary infiltrates, all features our patient exhibited. The CNS is usually spared; however, the retina’s involvement as an embryologic extension of the CNS shows this is not entirely true. Variability in the presence of retinal stigmata is understood to be secondary to differences in the ASM mutation in the SMPD1 gene. 5
In a review of 45 patients with Niemann-Pick disease type B, 12 patients (26.67%) had the perception of a cherry-red spot and 3 patients (6.67%) had bilateral symmetric macular halos. 5 Epigenetics may also play a role because siblings with the same mutation might not all develop retinal stigmata. 5 The cherry-red spots are likely a pseudoappearance and are different from those seen in central retinal artery occlusion (CRAO). In CRAO, cherry-red spots can occur because the associated retinal ischemia causes opacification of the vascularized inner retina but not of the foveal avascular zone (FAZ), which has a choroidal blood supply.
Macular halos are typically asymptomatic. 3 Multimodal imaging of macular halos in Niemann-Pick disease has enabled better characterization of the disease. The clinical examination or color fundus photography shows a white, crystalloid ring up to 3.0 mm in diameter encircling the unaffected fovea.5 –7 Lipid deposition is hypothesized to occur in retinal ganglion cells, which are not present in the fovea, thus giving rise to a fovea-sparing red spot.5,6 Macular halos have previously been reported to be hypoautofluorescent. 8 This differs from the halos in our case, in which the autofluorescence was unaffected in the area of the halo but a small foveal hyperautofluorescent dot was seen in the right fovea. Red-free photography shows annular hyperreflectivity, 2 as seen in our case.
On OCT, inner retinal granular hyperreflective deposits in the parafoveal region at the foveal slope (clivus) without disruption of the outer retinal layers are typically seen.1,6,7 Some authors have suggested its exclusive presence in the GCL, 6 and others have suggested involvement of the IPL and inner nuclear layer (INL). 7 Kim et al 6 supported the theory of GCL involvement given the absence of retinal ganglion cells in the fovea, which is usually not involved in the halo. On the contrary, Rudich et al 7 suggested that the macular halo is likely not limited to the retinal ganglion cells alone because the foveal slope also contains the termini of the IPL and INL. It was proposed that the hyperreflectivity could arise from foveal Müller glia, which span the entire retina and fan out radially in the inner retina. 7 Unique properties, including high metabolic activity, may make the Müller glia susceptible to sphingomyelin deposition.7,8 Bolukbasi et al 8 reported a case of foveal vertical hyperreflectivity extending from the ILM to the external limiting membrane (ELM), further supporting involvement of Müller cells.
Until a recent report of OCT angiography (OCTA) findings, the retinal and choroidal vasculature was thought to be unaffected in Niemann-Pick disease given unremarkable findings on fluorescein angiography.1,2 On OCTA, the presence of capillary flow voids in the superficial plexus with vascular tortuosity and changes in the FAZ suggest that vascular endothelial cells may be affected. 8
Limited electrophysiologic evidence of patients with Niemann-Pick disease type B suggests a possible functional impairment of the retina and optic nerve pathway.1,6 In a previous study, 6 multifocal electroretinography in a patient showed a localized depression in the central peak amplitude corresponding to the macular halo. In another study, 1 a patient was found to have reduced amplitude on full-field electroretinography, prolonged latencies on flash visual evoked potential, and a reduction in sensitivity on microperimetry.
It is likely that there is a spectrum of retinal layer involvement in patients with macular halos based on the reservoir of functional enzyme activity. In a case series by McGovern et al, 5 2 of 45 patients from different families with Niemann-Pick disease type B had unremarkable examinations until the ages of 7 years and 9 years, when the manifestation of macular halos was noted. This suggests that macular halos may develop or become more prominent over time.
Localization of halos is not well understood because of the limited histopathologic evidence. On light microscopy and electron microscopy of the globe of a 3-year-old girl with Niemann-Pick disease type A, Libert et al 9 described lipid-rich material organized into lamellar inclusion membranes in the retinal ganglion cells. In the case of a 2-year-old girl with Niemann-Pick disease type A, Robb and Kuwabara 4 described deposition in membranous cytoplasmic bodies that were present in the cornea, lens epithelium, iris sphincter, and retina. In addition to heavy deposition in the retinal ganglion cells, membranous cytoplasmic bodies were also found in amacrine cells, Müller cells, glial cells, and photoreceptors. The membranous cytoplasmic bodies were noted to assume a more regular appearance in retinal ganglion cells, whereas they appeared more irregular and vacuolated in Müller cells and retinal pigment epithelium. It is possible that the intracellular arrangement of these deposits determines the degree of hyperreflectivity on OCT. The uniform arrangement of deposits in retinal ganglion cells may generate a stronger reflectance signal in the GCL, while the scattered arrangement in other cells might result in a less intense signal, resulting in a false impression of exclusive GCL involvement. Histologic evidence of the involvement of other retinal cells in the case by Robb and Kuwabara 4 refutes the theory of exclusive lipid deposits in the GCL.
An opportunistic unremarkable cytopathologic analysis of the ILM suggests it is not involved in macular halos. This is consistent with the lack of ILM involvement found on the postmortem analysis of the patient with Niemann-Pick disease type A in the study by Libert et al. 9 The persistence of the macular halo in our patient after ILM peeling surgery further confirms that the ILM is not involved. The ILM is a basement membrane elaborated by Müller glia endfeet. Cytology confirmed typical appearances for ILM peeling specimens, including the ILM with scant attached inner neural retina without evidence of gliosis. This finding suggests that if lipid deposition involved Müller glia, it may be limited to the cell soma or apex. The absence of macrophages and microglia in the vitreous washings also argues against a reactive process at the vitreoretinal interface in our patient.
The concurrent presence of the FTMH and macular halo could be linked to an underlying dysfunction of Müller cells. OCT findings have suggested that FTMH formation is related to Müller cell integrity. 10 After the disruption of inner retinal layers, the Müller cell cone provides structural integrity to the outer retinal layers by integrating its processes with photoreceptor cells. 10 Closure of an FTMH can be mediated by the formation of a hyperreflective glial band at the ELM and the facilitation of centripetal photoreceptor displacement through the concentric contraction of its processes. 10 If the presence of lipid deposition alters the structure and function of the Müller cell cone to maintain the integrity of the outer retinal layers, the presence of the macular halo may have contributed to the FTMH formation in our patient.
Although potential Müller cell dysfunction in Niemann-Pick disease type A and type B provides a theoretical explanation for FTMH formation, it is also possible that the pathogenesis in our case was purely the result of vitreofoveal traction secondary to age-related vitreous synchysis and syneresis. In our patient, the OCT scans showed that the vitreous was lifted at the margin of the hole (Figure 1E). The appearance of the FTMH with hydration and cystic edema in a parafoveal location is typical of iatrogenic FTMHs. There were no atypical features, such as loss of retinal tissue seen in MHs associated with macular telangiectasia type II, or a giant MH, as seen in Alport disease. No abnormalities of the vitreoretinal interface were seen in the contralateral eye, nor have any been described in the literature to our knowledge.
In conclusion, the presence of macular halos should prompt an evaluation for Niemann-Pick disease. Multimodal imaging can assist in their identification. The management of an FTMH in the presence of macular halos is unchanged, with successful closure achieved by vitrectomy, ILM peeling, and gas tamponade. The etiology of the macular halo in Niemann-Pick disease type B remains unclear but likely involves sphingomyelin deposition at the foveal slope, possibly in retinal ganglion cells and Müller glia. The ILM appears to be uninvolved.
Footnotes
Ethical Approval
This case report did not require ethical approval.
Statement of Informed Consent
The patient consented to publication of the clinical history, findings, and images.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.