
Abstract
Purpose:
To compare intravitreal nesvacumab (anti-angiopoietin-2) + aflibercept vs intravitreal aflibercept injection (IAI) in neovascular age-related macular degeneration (nAMD).
Methods:
Eyes were randomized (1:2:3) to nesvacumab 3 mg + aflibercept 2 mg (LD combo), nesvacumab 6 mg + aflibercept 2 mg (HD combo), or IAI 2 mg at baseline, week 4, and week 8. The LD combo was continued every 8 weeks (q8w). At week 12, the HD combo was re-randomized to q8w or every 12 weeks (q12w) and IAI was re-randomized to q8w, q12w, or HD combo q8w through week 32.
Results:
The study comprised 365 eyes. At week 12, the mean best-corrected visual acuity (BCVA) gains from baseline were similar in the LD combo group, HD combo group, and IAI group (5.2 letters, 5.6 letters, and 5.4 letters, respectively); the mean central subfield thickness (CST) reductions were similar (182.2 µm, 200.0 µm, and 178.6 µm, respectively). The mean changes in BCVA and CST through week 36 were similar across groups. At week 12, complete retinal fluid resolution was observed in 49.1% (LD combo), 50.8% (HD combo), and 43.6% (IAI) of eyes; the proportions with a CST of 300 μm or less were similar across groups. Numerical trends at week 32 toward complete retinal fluid resolution with combination treatment were not maintained at week 36. Serious ocular adverse events were infrequent and comparable across groups.
Conclusions:
In nAMD, nesvacumab + aflibercept showed no additional BCVA or CST benefit over IAI monotherapy.
Keywords
Introduction
The current standard of care for neovascular age-related macular degeneration (nAMD) is treatment with antivascular endothelial growth factor (VEGF) agents, including intravitreal aflibercept injection (IAI) and ranibizumab.1,2 However, not all patients with nAMD regain visual acuity (VA) to 20/40 or better after anti-VEGF therapy.
There may be an opportunity for further improvement and the possibility of lessening the treatment burden by further extending the dosing interval.3–5 One potential way this might be achieved is through combination treatment targeting other mechanisms of action involved in the pathogenesis of nAMD. Studies involving preclinical models of pathologic ocular angiogenesis suggest that VEGF and angiopoietin-2 (Ang2) are co-regulated in nAMD and may work together to promote pathologic neovascularization and increase vascular permeability.6–8
Intravitreal nesvacumab + aflibercept fixed-dose combination therapy provides specific doses of the anti-Ang2 antibody with a known effective dose of the anti-VEGF fusion protein, aflibercept 2 mg. This dual mechanism approach is also being investigated with the bispecific antibody faricimab. 9 The fixed-dose combination may have advantages in that both antibodies can be individually optimized, ensuring a specific dose of each component is delivered. A bispecific antibody does not allow flexibility to dose at different ratios, and binding is limited to a single ligand—either VEGF or Ang2—per molecule.
The phase 2 ONYX trial was undertaken to compare the efficacy and safety of intravitreal nesvacumab + aflibercept fixed-dose combination vs IAI monotherapy in patients with nAMD.
Methods
Study Design
ONYX
Eligible patients were treatment-naive, aged 50 years or older, had active subfoveal choroidal neovascularization (CNV) secondary to nAMD (including juxtafoveal lesions affecting the fovea as evidenced by fluorescein angiography [FA] in the study eye assessed by the central reading center), and had a best-corrected VA (BCVA) score of 73 to 24 Early Treatment Diabetic Retinopathy Study (ETDRS) letters (20/40 to 20/320 Snellen equivalent) in the study eye. Key exclusion criteria included evidence of CNV from any cause other than AMD in either eye, previous treatment with an anti-VEGF agent in the study eye, evidence of diabetic macular edema (DME) or diabetic retinopathy (DR) in either eye, or any history of stage 2 or greater macular hole in the study eye.
One eye per eligible patient was randomized (1:2:3) via an interactive voice/web response system to receive intravitreal low-dose nesvacumab 3 mg + aflibercept 2 mg (LD combo), high-dose nesvacumab 6 mg + aflibercept 2 mg (HD combo), or IAI 2 mg at baseline, week 4, and week 8. The Supplemental Methods provides information on the nesvacumab + aflibercept drug formulation. The LD combo treatment continued every 8 weeks (q8w) from week 16 to week 32. The HD combo group was re-randomized to HD combo q8w from week 16 or every 12 weeks (q12w) from week 20 until week 32. The IAI group was re-randomized to IAI q8w from week 16, IAI q12w from week 20, or HD combo q8w (IAI monotherapy → HD combo q8w) from week 16 through week 32 (Figure 1).
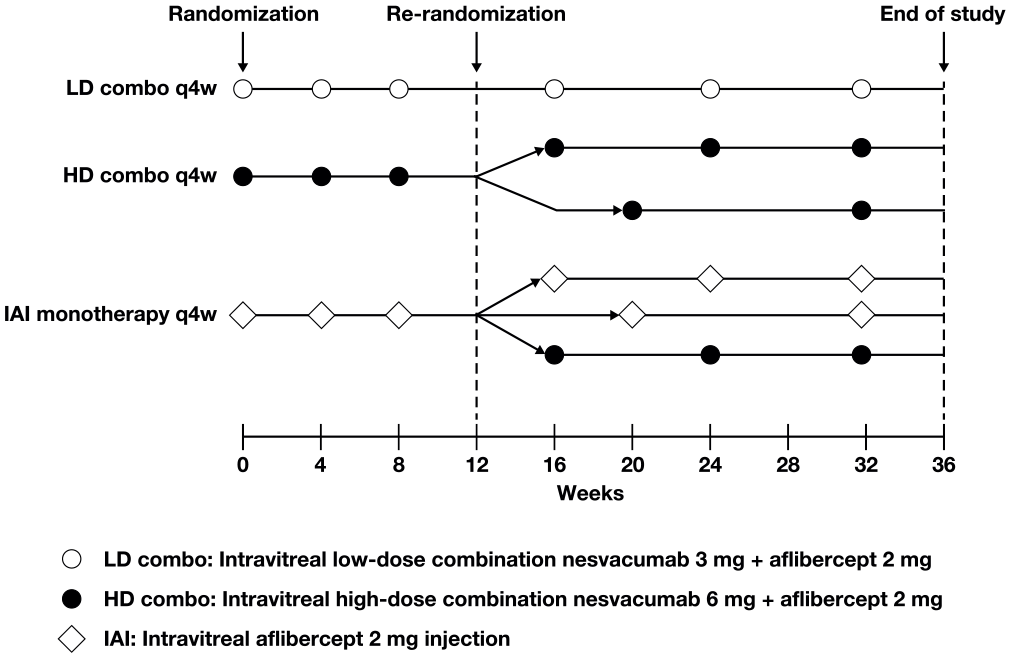
ONYX trial design. Eyes receiving LD combo continued treatment q8w. At week 12, eyes receiving HD combo were re-randomized to treatment q8w or q12w, and eyes receiving IAI were re-randomized to IAI q8w or q12w or HD combo q8w through week 32. Markers indicate when a dose of the respective treatments was administered.
Starting at week 12, additional treatment with IAI 2 mg could be administered if the study eye did not respond to the protocol-specified dosing interval as a result of persistent or worsening disease as assessed by the investigator. Patients requiring additional interim treatment continued to receive randomized treatment at subsequent visits and remained masked to the dosing interval.
Outcome Measures
The primary efficacy endpoint was the change in BCVA from baseline (in ETDRS letters) at week 12 and week 36. Secondary efficacy endpoints were the change from baseline in central subfield thickness (CST) assessed by spectral-domain optical coherence tomography (SD-OCT), CNV area by FA, and total lesion area by FA at weeks 12 and 36. The change in CST was assessed by SD-OCT, CNV area by FA, and total lesion area by FA. Other efficacy endpoints included resolution of retinal fluid (subretinal and intraretinal) on SD-OCT and normalization of macular thickness (CST ≤300 μm) at week 12 and week 36. The Spectralis (Heidelberg Engineering, Inc) and Cirrus (Carl Zeiss Meditec, Inc) were used for SD-OCT; the same SD-OCT machine was used for each patient throughout the study. The central reading center (Duke Reading Center, Durham, NC, USA) provided comparable measurements of CST for both SD-OCT platforms.
Systemic safety was assessed by adverse events (AEs), vital signs, electrocardiograms, and laboratory tests. Ocular safety was assessed by ophthalmic examinations, including intraocular pressure measurement, slitlamp examination, and indirect ophthalmoscopy. The BCVA, SD-OCT, and safety evaluations were performed at each study visit, which occurred every 4 weeks.
Statistical Analysis
Assuming a normal distribution for the change in BCVA from baseline to week 12, a mean between-group difference of 5 letters (SD, 11 letters) and a dropout rate of 15%, a sample size of 60 patients, 120 patients, and 180 patients in the LD combo group, HD combo group, and IAI group, respectively, was needed to provide an 80% or greater probability that the 95% CI for treatment difference would exclude zero. The sample-size calculation was computed using the 2-group Satterthwaite 10 t test of unequal sample size with a 1:2:3 ratio by clinical assumption with equal variances using nQuery Advisor 7.0 software (Statsols).
The full analysis set, which included all randomized patients who received any study treatment and had a baseline BCVA measurement and 1 or more post-baseline BCVA measurements was used to analyze all efficacy variables through week 12. The secondary randomization set included all patients in the full analysis set who completed the trial through week 12, had received any study treatment after week 12 (patients originally randomized to LD combo) or after re-randomization (patients originally randomized to HD combo or IAI), and had a BCVA measurement at week 12 and 1 or more BCVA measurements after week 16. The secondary randomization set was used to analyze all efficacy variables through week 36. The assessment of outcomes from week 12 through week 36 included a qualitative assessment of effect over these visits to determine the overall interpretation of benefit.
The primary efficacy endpoint at week 12 was analyzed using an analysis of covariance (ANCOVA) model, with treatment as the main effect and baseline BCVA as the covariate. The least-square (LS) mean and 2-sided 95% CI of the difference (LD or HD combo minus IAI) were calculated. After week 12, the primary efficacy endpoint was analyzed using an ANCOVA model with baseline BCVA as the covariate and treatment group and BCVA stratification variable as fixed factors. Secondary efficacy endpoints were analyzed as for the primary efficacy endpoint. Proportions of eyes with resolution of retinal fluid and normalization of macular thickness were summarized using descriptive statistics. No formal statistical testing was performed; all reported P values are nominal. Nominal P values for comparison of either LD or HD combo with IAI were calculated using the 2-sided Cochran-Mantel-Haenszel test adjusted by the BCVA stratification variable. For efficacy analyses, missing data were imputed using the last observation carried forward.
The safety analysis set, which included all patients who received 1 or more doses of any study drug, was used to analyze safety through week 36. Safety data were summarized using descriptive statistics.
Results
Patients
A total of 365 eyes were randomized to the LD combo group (n = 60), the HD combo group (n = 122), or the IAI group (n = 183). Supplemental Figure 1 shows the patient dispositions. In general, the baseline characteristics were balanced across the 3 treatment groups (Table 1); the mean age was 78.4 to 79.4 years, the mean BCVA was 55.5 to 58.2 letters, and the mean CST was 490.4 to 521.1 µm. Supplemental Table 2 shows the baseline characteristics of the secondary randomization set. Completion rates through week 36 across the 6 groups in the secondary randomization set were 93.0% to 98.3%.
Baseline Characteristics by Original Treatment Randomization. a
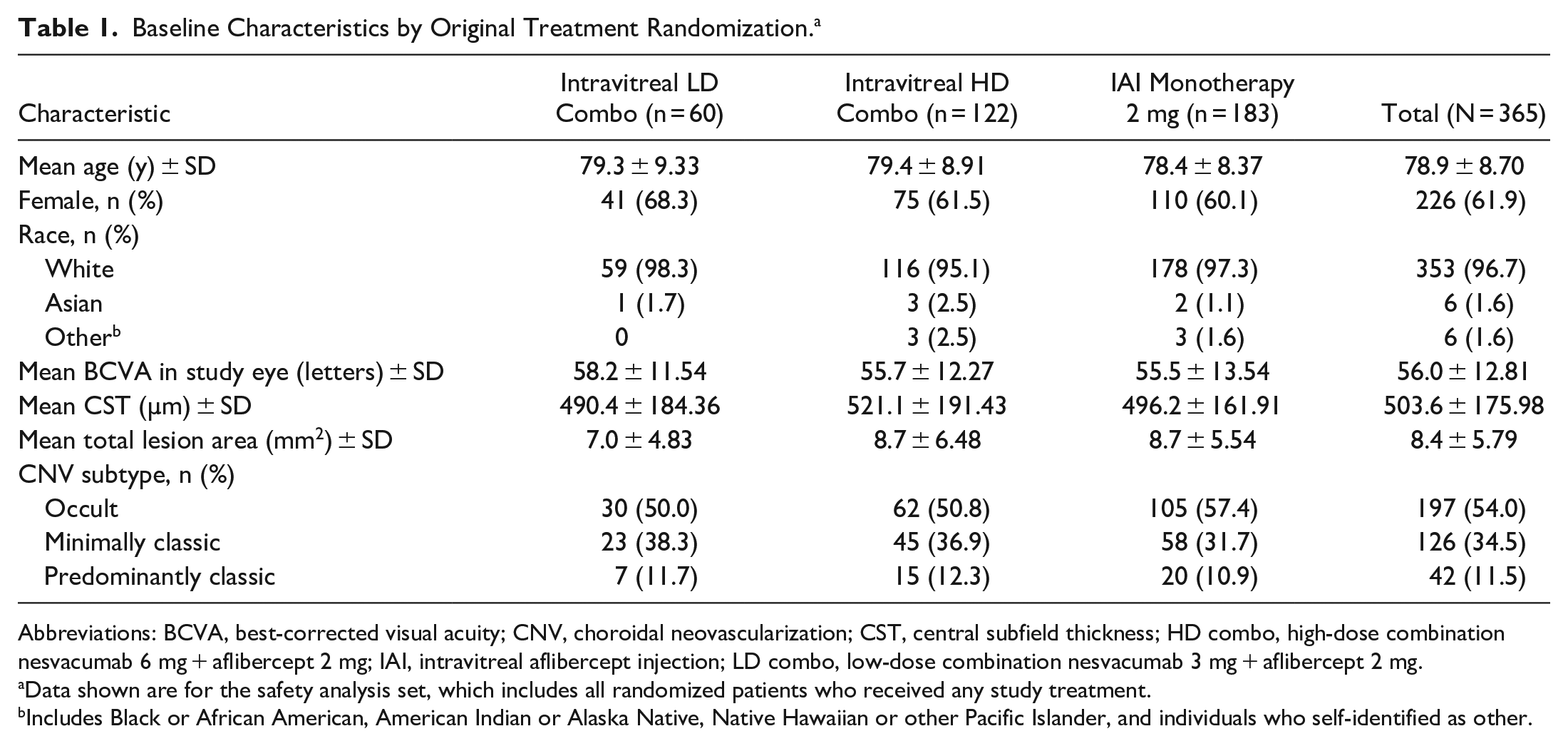
Abbreviations: BCVA, best-corrected visual acuity; CNV, choroidal neovascularization; CST, central subfield thickness; HD combo, high-dose combination nesvacumab 6 mg + aflibercept 2 mg; IAI, intravitreal aflibercept injection; LD combo, low-dose combination nesvacumab 3 mg + aflibercept 2 mg.
Data shown are for the safety analysis set, which includes all randomized patients who received any study treatment.
Includes Black or African American, American Indian or Alaska Native, Native Hawaiian or other Pacific Islander, and individuals who self-identified as other.
Over the 32-week treatment period, the mean ± SD number of intravitreal injections administered was 6.6 ± 0.93 in the LD combo q8w group, 5.9 ± 0.44 in the HD combo q8w group, 5.1 ± 0.45 in the HD combo q12w group, 5.9 ± 0.35 in the IAI q8w group, 4.9 ± 0.27 in the IAI q12w group, and 5.9 ± 0.40 in the IAI monotherapy → HD combo q8w group. A programming error in the interactive voice/web response system affected the dosing schedules for LD combo q8w and HD combo q12w, resulting in a reduced proportion of eyes being dosed per protocol after re-randomization. (Patients received additional doses or the doses were not timed as intended.) Approximately 50% and 75% of eyes were dosed per protocol in the LD combo q8w group and HD combo q12w group, respectively. Treatment schedules for all other groups were unaffected. The Supplemental Methods shows the details on this operational error and the patients receiving additional treatment.
Efficacy
The mean change in BCVA from baseline to week 12 in the LD and HD combo groups was 5.2 letters and 5.6 letters, respectively, vs 5.4 letters in the IAI group, with LS mean differences of 0.02 (95% CI −2.99 to 3.03; nominal P = .9894) between the LD combo group and IAI group and 0.25 (95% CI −2.09 to 2.59; nominal P = .8346) between the HD combo group and IAI group (Figure 2A). The mean change from baseline in BCVA at week 36 was 5.9 letters, 6.0 letters, 6.4 letters, 6.9 letters, 4.2 letters, and 2.9 letters in the LD combo q8w group, the HD combo q8w group, the HD combo q12w group, the IAI q8w group, the IAI q12w group, and the IAI monotherapy → HD combo q8w group, respectively (Figure 3A).
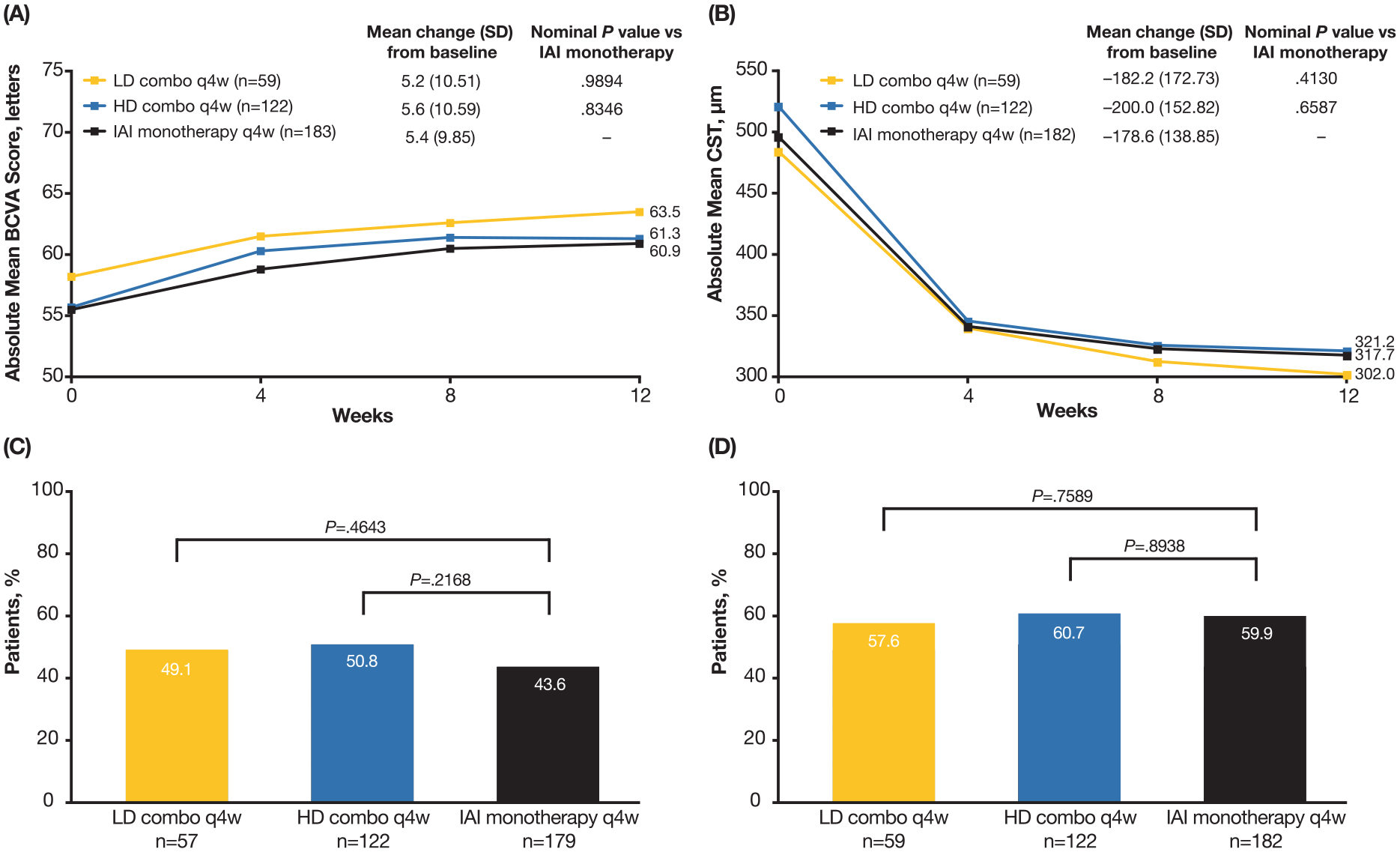
Visual and anatomic outcomes through week 12 by original treatment randomization. (A) Absolute mean BCVA score from baseline through week 12. (B) Absolute mean CST from baseline through week 12. (C) Proportion of eyes with complete resolution of retinal (subretinal and intraretinal) fluid at week 12. (D) Proportion of eyes with normalization of macular thickness (CST ≤300 μm) at week 12.
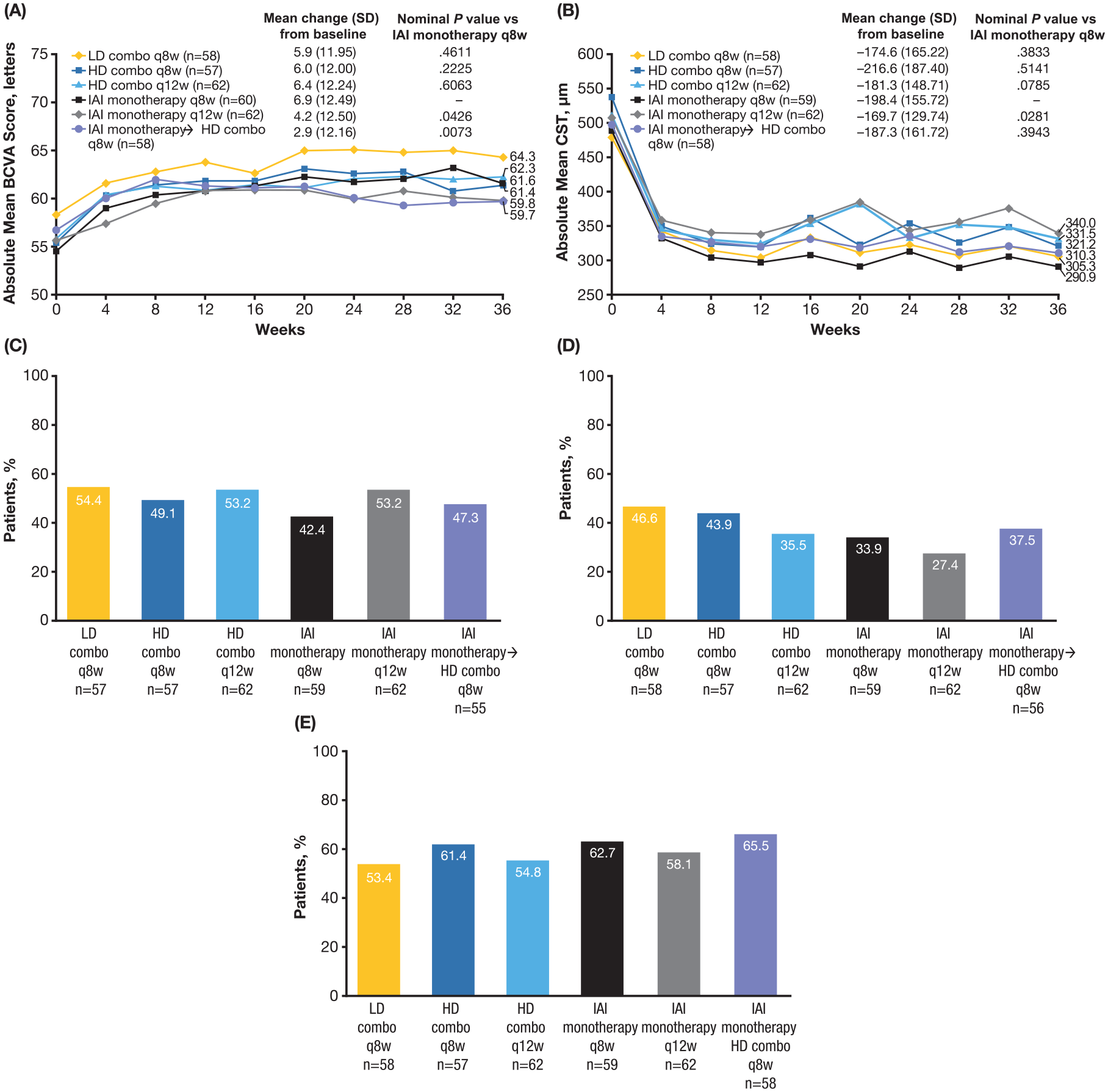
Visual and anatomic outcomes through week 36 by treatment assignment at week 12. (A) Absolute mean BCVA score from baseline through week 36. (B) Absolute mean CST from baseline through week 36. (C) Proportion of eyes with complete resolution of retinal fluid at week 36. (D) Proportion of eyes with complete resolution of retinal fluid at week 32. (E) Proportion of eyes with normalization of macular thickness (CST ≤300 μm) at week 36.
The mean change from baseline in CST at week 12 in the LD and HD combo groups was −182.2 µm and −200.0 µm, respectively, vs −178.6 µm in the IAI group (LD combo vs IAI, nominal P = .4130; HD combo vs IAI, nominal P = .6587) (Figure 2B). The mean change from baseline in CST at week 36 was −174.6 µm, −216.6 µm, −181.3 µm, −198.4 µm, −169.7 µm, and −187.3 µm, in the LD combo q8w group, the HD combo q8w group, the HD combo q12w group, the IAI q8w group, the IAI q12w group, and the IAI monotherapy → HD combo q8w group, respectively (Figure 3B). Data for changes from baseline in CNV area and total lesion area for the 3 original randomization groups and the secondary randomization set are described in Supplemental Table 3 and Supplemental Figure 2, respectively.
The proportion of eyes with complete resolution of retinal fluid at week 12 in the LD and HD combo groups was 49.1% and 50.8%, respectively, vs 43.6% in the IAI group (LD combo vs. IAI, nominal P = .4643; HD combo vs IAI, nominal P = .2168) (Figure 2C). The proportions were similar at week 36 (Figure 3C). At week 32 (corresponding to 8 weeks or 12 weeks since the last treatment and thus the trough of the dosing interval), the proportion of eyes with complete resolution of retinal fluid ranged from 27.4% to 46.6% (Figure 3D), with the largest proportion in the LD combo q8w group and the smallest proportion in the IAI q12w group.
The proportion of eyes with normalization of macular thickness (CST ≤300 μm) at week 12 in the LD and HD combo groups was 57.6% and 60.7%, respectively, vs 59.9% in the IAI group (LD combo vs IAI, nominal P = .7589; HD combo vs IAI, nominal P = .8938) (Figure 2D; week 36 results shown in Figure 3E).
Safety
Throughout the 36-week trial, 30.0% of patients (18/60), 42.6% of patients (52/122), and 29.0% of patients (53/183) in the LD combo group, HD combo group, and IAI group, respectively, had 1 or more ocular AEs in the study eye, most commonly conjunctival hemorrhage (LD combo, 3.3% [2/60]; HD combo, 4.9% [6/122]; IAI monotherapy, 2.7% [5/183]) (Table 2). The IAI group included 58 patients who received HD combo after re-randomization. Supplemental Table 4 shows the incidence of intraocular inflammation (iritis and iridocyclitis). No cases of vasculitis, intermediate uveitis, or posterior uveitis were reported. Serious ocular AEs occurred in the study eye of 3.3% of patients (2/60), 2.5% of patients (3/122), and 0% of patients (0/183) in the LD combo group, HD combo group, and IAI group, respectively. The only serious ocular AE that occurred in more than 1 patient was reduced VA, which occurred in 2 patients; 1 of these 2 events was considered related to the study treatment.
Safety Through Week 36 by Original Treatment Randomization. a
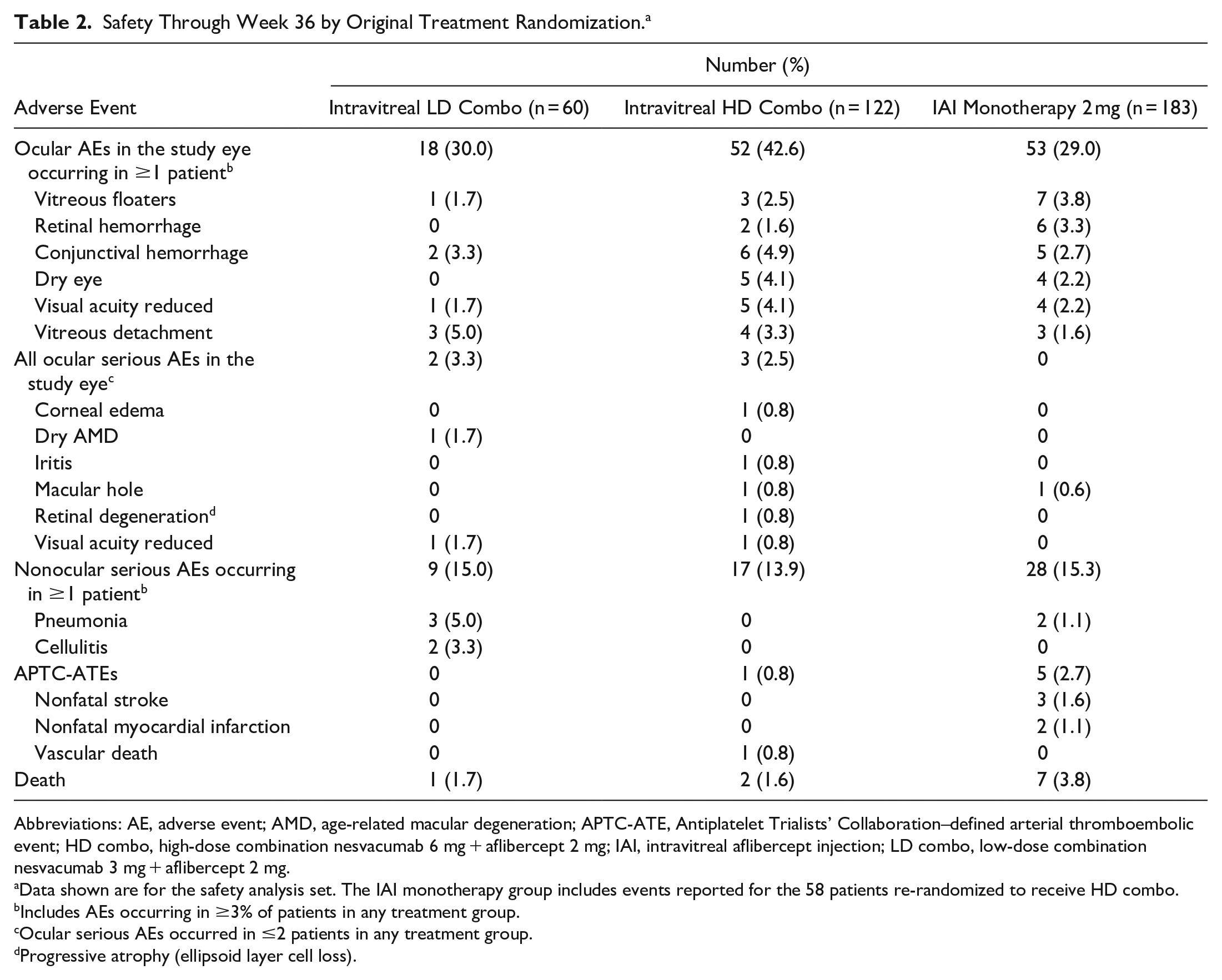
Abbreviations: AE, adverse event; AMD, age-related macular degeneration; APTC-ATE, Antiplatelet Trialists’ Collaboration–defined arterial thromboembolic event; HD combo, high-dose combination nesvacumab 6 mg + aflibercept 2 mg; IAI, intravitreal aflibercept injection; LD combo, low-dose combination nesvacumab 3 mg + aflibercept 2 mg.
Data shown are for the safety analysis set. The IAI monotherapy group includes events reported for the 58 patients re-randomized to receive HD combo.
Includes AEs occurring in ≥3% of patients in any treatment group.
Ocular serious AEs occurred in ≤2 patients in any treatment group.
Progressive atrophy (ellipsoid layer cell loss).
Serious nonocular AEs occurred in 15.0% of patients (9/60), 13.9% of patients (17/122), and 15.3% of patients (28/183) in the LD combo group, HD combo group, and IAI group, respectively (Table 2), most commonly pneumonia (LD combo, 5.0% [3/60]; HD combo, 0% [0/122]; IAI monotherapy, 1.1% [2/183]) and a fall (LD combo, 1.7% [1/60]; HD combo, 1.6% [2/122]; IAI monotherapy, 0.5% [1/183]). Antiplatelet Trialists’ Collaboration (APTC)–defined arterial thromboembolic events (myocardial infarction, stroke, or death from vascular or unknown causes) occurred in 0% of patients (0/60), 0.8% of patients (1/122), and 2.7% of patients (5/183) in the LD combo group, HD combo group, and IAI group, respectively. Ten patients died during the study (LD combo, n = 1; HD combo, n = 2; IAI, n = 7); no death was considered related to the study treatment. Supplemental Table 5 shows the safety data for the secondary randomization.
Conclusions
In the primary efficacy analysis of this exploratory phase 2 study, dual inhibition of Ang2 and VEGF with a fixed-dose combination of intravitreal nesvacumab + aflibercept (either LD or HD) in patients with treatment-naive nAMD did not lead to improvements in the absolute mean BCVA score through week 12 and week 36 relative to IAI monotherapy. Nesvacumab + aflibercept produced similar outcomes in patients with DME in the RUBY trial. 11
The efficacy of IAI monotherapy, as a control treatment, was consistent with results in previous studies of nAMD.5,12 In general, both the IAI q8w group and q12w group maintained their VA gains at week 12 through week 36. The absolute mean changes in CST between baseline and week 12 were also similar across treatment groups. The mean changes in BCVA and CST between baseline and week 36 were consistent with results observed through week 12. In general, vision and anatomic responses were stable in patients switched from IAI every 4 weeks for 3 initial monthly doses to IAI q8w or q12w.
Qualitative grading of the resolution of retinal (subretinal and intraretinal) fluid showed some numeric trends in favor of the combination treatment; more eyes randomized to LD or HD combo had complete resolution of fluid at week 12 compared with IAI. This difference was especially notable at week 32, which was the end of the dosing interval (8 or 12 weeks since last treatment) in all groups. At week 36, although a greater percentage of eyes assigned to LD combo q8w and HD combo q8w had complete resolution of fluid than those assigned to IAI q8w, these proportions were comparable to the proportion in the IAI q12w group. No added benefit from week 12 to week 36 was observed in the IAI monotherapy → HD combo q8w group.
No positive trend in favor of combination treatment was seen for the proportion of eyes achieving a “normal” macular thickness (CST ≤300 µm), a reduction in CNV, or total lesion area. Although these anatomic changes could signal some additional benefit from combination therapy, they did not translate into clinically or statistically significant improvements in VA. Interpretation of these assessments is limited by the small samples, particularly after re-randomization, and the relatively short time frame (36 weeks) of the study. Whether a longer duration of Ang2 inhibition will result in improved clinical outcomes, such as decreased fibrosis or improved long-term durability, requires further study.
Although Ang2 is upregulated in patients with nAMD and plays a role in pathologic neovascularization, 13 its importance is unclear. In comparison, VEGF appears to play a major role in neovascularization based on the clinical efficacy of anti-VEGF agents, including IAI monotherapy.12,14 In vitreous samples collected from patients newly diagnosed with nAMD, retinal vein occlusion, DR, or proliferative DR, Ang2 levels were elevated compared with those in control patients diagnosed with a macular hole. 15 Across these 4 retinal vascular diseases, the levels of Ang2 in patients with nAMD were the least elevated; however, it has also been shown that patients with nAMD have lower aqueous VEGF levels than patients with diabetic eye disease, 16 highlighting that the interaction among various mechanisms is not yet fully understood.
The mechanism by which VEGF induces neovascularization and leakage is better understood than that for Ang2, which situationally acts as both an agonist and antagonist to its receptor, tyrosine kinase. Although there is preclinical evidence for the involvement of both Ang2 and VEGF in the pathogenesis of nAMD and a synergistic effect in animal models of disease, 17 findings from the ONYX clinical trial did not provide evidence that combined VEGF and Ang2 blockade improved visual outcomes, although there was a trend toward some improved anatomic outcomes.
The most frequently observed ocular AEs were consistent with those experienced after intravitreal injections, nAMD, and the elderly patient population.4,12,18 The frequency of serious ocular AEs was higher in the LD combo group and HD combo group than in the IAI group; however, the rate of serious ocular AEs was low across all groups, with each event occurring in 2 or fewer patients. The incidence of APTC-defined arterial thromboembolic events and death was low during the 36-week study. Overall, the safety profile of intravitreal aflibercept + nesvacumab was similar to that of IAI monotherapy, with no new or unexpected safety signals.
Limitations of this study include the relatively small sample, particularly after the week 12 re-randomization. Consistent with the exploratory nature of this study, there was no correction for multiplicity; thus, all P values were considered nominal. Dosing errors affecting the LD combo q8w group and HD combo q12w group made interpretation of any dosing interval benefit difficult, although the mean number of doses received in those groups was not very different from the intended number of doses.
The ONYX trial did not show additional benefits in VA or CST reductions with intravitreal nesvacumab + aflibercept combination treatment over IAI monotherapy in treatment-naive nAMD at 36 weeks, although there were some numeric trends toward greater anatomic effects (eg, retinal dryness), which may warrant further investigation of the role of anti-Ang2 in combination with anti-VEGF.
Supplemental Material
sj-docx-1-vrd-10.1177_24741264221126061 – Supplemental material for Intravitreal Nesvacumab (Anti-Angiopoietin-2) Plus Aflibercept in Neovascular AMD: Phase 2 ONYX Randomized Trial
Supplemental material, sj-docx-1-vrd-10.1177_24741264221126061 for Intravitreal Nesvacumab (Anti-Angiopoietin-2) Plus Aflibercept in Neovascular AMD: Phase 2 ONYX Randomized Trial by Jeffrey S. Heier, Allen C. Ho, David S. Boyer, Karl Csaky, Robert Vitti, Lorah Perlee, Karen W. Chu, Friedrich Asmus, Sergio Leal, Oliver Zeitz, Yenchieh Cheng, Thomas Schmelter and David M. Brown in Journal of VitreoRetinal Diseases
Footnotes
Acknowledgements
The authors thank the study investigators and patients involved in the study. Medical writing support was provided by Melissa Purves, PhD, and Nila Bhana, MSc, of Prime (Knutsford, United Kingdom), according to Good Publication Practice guidelines and was funded by Regeneron Pharmaceuticals, Inc. The authors were involved in the study design and collection, analysis, and interpretation of data. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Ethical Approval
ONYX (NCT02713204) was conducted across 77 sites in the United States, in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guideline for Good Clinical Practice, between March 2016 and October 2017. Approvals were obtained from Institutional Review Boards/Ethics Committees at each study site.
Statement of Informed Consent
All patients provided written informed consent prior to study commencement.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Jeffrey S. Heier reports consulting for 4DMT, Adverum, Aerie, Aerpio, Aldeyra, Allegro, Alzheon, Annexon, Apellis, Aprea, Asclepix, Aviceda, Beaver-Visitec, Bionic Vision Technologies, DTx, Eloxx, Galimedix, Genentech, Graybug, Gyroscope, Horizon Therapeutics, iRenix, Iveric, JCyte, Chengdu Kanghong Biotechnology, LensGen, NGM, Notal Vision, Novartis, Ocular Therapeutix, OccuTerra, Oxurion, Palatin, Regeneron Pharmaceuticals, Inc, RegenXBio, Stealth, Thea, Verseon, Vinci, and Voyant; and research grants from Aerpio, Aldeyra, Apellis, Asclepix, Bayer, Genentech, Graybug, Gyroscope, Hermera, Iveric, Janssen R&D, JCyte, Chengdu Kanghong Biotechnology, Kodiak, Novartis, Regeneron Pharmaceuticals, Inc, RegenXBio, and Stealth.
Allen C. Ho reports consulting for and receiving research grant support from Adverum, Aerie, AGTC, Alcon Laboratories, Inc, Aldeyra, Allergan, Apellis, Asclepix, Atsena, Beaver-Visitec International, Inc, Chengdu Kanghong Biotechnology, Clearside, Covalent Medical, Genentech, Graybug, Gyroscope, Iveric, Janssen/Johnson & Johnson, Lineage, Lumithera, National Eye Institute, Notal, Novartis, Ocular Therapeutics, ONL, ProQR, Regeneron Pharmaceuticals, Inc, and RegenXBio.
David S. Boyer reports consulting for Genentech, Regeneron Pharmaceuticals, Inc, Roche, Bayer, Allergan, Chengdu Kanghong Biotechnology, Clearside Biomedical, Boehringer Ingelheim, Kodiak, Novartis, Adverum, RegenXBio, and Graybug.
Karl Csaky reports consulting for Genentech, Roche, Allergan, Novartis, and Regeneron Pharmaceuticals, Inc.
Robert Vitti, Lorah Perlee, Karen W. Chu, and Yenchieh Cheng are employees of and stockholders in Regeneron Pharmaceuticals, Inc.
Friedrich Asmus was an employee of Bayer AG at the time of the study.
Sergio Leal is an employee of and stockholder in Bayer Consumer Care AG.
Oliver Zeitz reports consulting and serving as a speaker for Allergan, Bayer, Boehringer-Ingelheim, and Novartis; consulting for Omeicos and Oxular; and research support from Bayer, Boehringer-Ingelheim, and Novartis. He was an employee of Bayer until September 30, 2016.
Thomas Schmelter is an employee of and stockholder in Bayer AG.
David M. Brown reports consulting for and receiving research funding from Regeneron Pharmaceuticals, Inc, Genentech/Roche, Novartis, Heidelberg, Allergan, Chengdu Kanghong Biotechnology, Santen, OHR, Clearside Biomedical, Adverum, RegenXBio, and Samsung; and consulting for Bayer, Optos, Kodiak, Senju, and Biotime, outside of the submitted work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The ONYX trial was funded by Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA. The sponsor participated in the design and conduct of the trial, analysis of the data, and preparation of the manuscript.
Data Sharing Statement
Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the indication has been approved by major health authorities, if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Submit requests to ![]() .
.
Supplemental Material
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.