
Abstract
Ciprofloxacin (CIP), a widely used antibiotic, is a poor biopharmaceutical resulting in low bioavailability. We optimized a CIP polymer–lipid hybrid nanoparticle (CIP-PLN) delivery system to enhance its biopharmaceutical attributes and the overall therapeutic performance. CIP-PLN formulations were prepared by a direct emulsification–solvent–evaporation method. Varying the type and ratio of lipid was tried to optimize a CIP-PLN formulation. All the prepared formulations were evaluated for their particle size, polydispersity index, zeta potential, physical stability, and drug entrapment efficiency. The drug in vitro release profile was also studied. Antibacterial activities were tested by the agar diffusion method for all CIP-PLN formulations against an Escherichia coli clinical bacterial isolate (EC04). CIP-PLN formulations showed average sizes in the range of 133.9 ± 1.7 nm to 217.1 ± 0.8 nm, exhibiting high size uniformity as indicated by polydispersity indices lower than 0.25. The entrapment efficiency was close to 80% for all formulations. The differential scanning calorimetry (DSC) thermograms indicated the existence of CIP in the amorphous state in all PLN formulations. Fourier transform infrared spectra indicated deep incorporation of molecular CIP within the polymer matrix. The release profile of CIP from PLN formulas showed a uniform prolonged drug profile, extended for a week from most formulations with a zero-order kinetics. The antibacterial activity of CIP-PLN formulations showed significantly higher antibacterial activity only with F4 containing lecithin as the lipid component. In conclusion, we successfully optimized a CIP-PLN formulation with a low nanoparticle size in a close range, high percentage of entrapment efficiency and drug loading, uniform prolonged release rate, and higher antibacterial activity against the EC04 clinical isolate.
Introduction
The applications of nanotechnology have tremendously emerged in both the therapeutic and diagnostic fields as promising tools to enhance the outcomes for many intractable diseases, including cancer and cardiovascular and neurodegenerative diseases. 1 The ultrasmall volume and the relatively high surface area have enabled nanoparticles to exhibit a wider distribution and higher concentration at the site of action and have allowed for avoidance of clearance by the reticuloendothelial system.2,3 The unique phospholipid bilayer nature and the phenomenal shell structure similar to that of biological cell membranes are considered important advantages of liposomes over many other classes of nanoparticulate systems due to their higher biocompatibility and nonimmunogenicity.4,5 However, they have poor physical and chemical stability characteristics, resulting in immature drug leakage and variability in biological drug release and distribution. 6 In addition, liposomal technology is insufficiently suited for the pharmaceutical industry due to its high production cost and batch-to-batch manufacturing variability.7,8
Solid lipid nanoparticles (SLNs) are a type of lipid-based matrix nanoparticles made from lipids that remain solid above body temperature. 9 Interestingly, SLNs were introduced in the 1990s as a potential alternative to overcome both the instability problems of liposomes and their high production cost while sharing the high advantage of their biocompatibility.10,11 SLNs carry some drawbacks, including a high initial drug release and modest stability in a tropical environment.11–13
Polymeric nanoparticles are matrix particles with a high encapsulating capacity of drugs regardless of their polarity. They also have high resistance to degradation and a uniform controlled release profile; they can cause local toxicities and irritations. 14
Polymer–lipid hybrid nanoparticles (PLNs) have been recently introduced as a new generation of nanoparticulate drug delivery systems to exploit the features of polymeric nanoparticles, such as higher structural stability and controlling release properties, with the higher biocompatibility and cellular uptake abilities of lipid nanoparticles.15,16
The architecture design of PLN can be described as a polymeric core layer in which the drug is homogeneously distributed, surrounded by a phospholipid coat layer with or without the presence of polyethylene glycol (PEG); therefore, it is called a lipid–polymer hybrid nanoparticle (LPN). 16 Other reports have described different configurations of a PLN system, where therapeutic moieties can be dispersed in an SLN core layer and surrounded by a hydrophilic polymer coat layer. 17 Also, therapeutic moieties can be dispersed homogeneously alongside the polymer in the solid lipid matrix.18,19
PLN formulations have reportedly shown enhancement of the efficacy of some anticancer drugs by reducing multidrug resistance mechanism effects, which has resulted in a huge reduction in the systemic toxicity.20,21
Overall, coupling different lipids and polymers that have different physical and chemical properties together in PLN formulation would provide promising alternatives to fit the delivery needs of diverse drugs and different drug combinations. 22
Ciprofloxacin (CIP) is a well-known quinolone antibiotic indicated for the treatment of respiratory tract infections, urinary tract infections, acute or chronic prostatitis, and gonorrhea.
23
According to the biopharmaceutical classification system (BCS), CIP is classified as class 4, which has thus made its use unfavorable for the oral route of administration due to its low permeability and low solubility. CIP has a low bioavailability, which requires the use of high doses resulting in a higher profile of adverse effects.
24
This has made CIP a potential candidate to be incorporated into many nanoparticulate drug delivery systems, including SLN, poly(
The aim of this study is to optimize a polymer–lipid hybrid nanoparticulate system for enhancing the therapeutic performance of CIP.
Materials and Methods
Materials
CIP was a generous gift from Tabuk Pharmaceuticals (Tabuk, Saudi Arabia). Imwitor 900K and Dynasan 118 were purchased from Sasol Germany GmbH (Witten, Germany). Mueller Hinton Agar was obtained from HiMedia Laboratories Pvt. Ltd. (Mumbai, India). Stearic acid, Tween 80, lecithin, and sodium deoxycholate were bought from Sigma-Aldrich Chemical Co. (St. Louis, MO). All the other materials were analytical grade.
Drug Analysis
CIP was analyzed by simple ultraviolet (UV) spectrophotometric assay method. A set of CIP standard solutions in methanol with various concentrations were prepared and their UV absorbance was measured at λ = 277 nm using a Thermo Scientific Evolution 60S UV-Visible Spectrophotometer (Thermo Scientific, Shanghai, China). The UV absorbance data were plotted against their corresponding concentrations to develop a standard calibration curve. The developed standard curve showed very good linearity for a wide range of concentrations from 1 to 20 µg/mL with R 2 = 0.999.
Preparation of CIP-Loaded Polycaprolactone–Lipid Hybrid Nanoparticles
Certain weights of polycaprolactone (PCL), lipid components, and 10 mg of CIP were dissolved in chloroform. An aqueous surfactant solution containing 10 mg/mL Tween 80 and 5 mg/mL sodium deoxycholate was prepared. Five milliliters of the chloroform solution was emulsified in 10 mL of the surfactant solution using probe sonication for 3 min at 40% intensity under ice. The formed emulsion was immediately redispersed in another 20 mL of the surfactant solution by applying another 2 min probe sonication under the same condition. The dispersion was stirred at room temperature under a hood for 5 h to allow the removal of chloroform. The formed nanoparticles were separated by centrifugation for 30 min using 15,000 relative centrifugal force (RCF), and the residue was washed twice and redispersed in distilled water and centrifuged. The washed particles were freeze-dried for 48 h using Christ Beta 2-8 LD Plus (Martin Christ, Germany). For optimization purposes, the composition of PLN formulations was varied to assess the effect of the type of lipid, the lipid-to-polymer ratio, and the drug-to-polymer/lipid ratio. Table 1 shows the exact composition of all the prepared formulas.
Composition of Each of the Prepared CIP-Polycaprolactone–Lipid Hybrid Nanoparticle Formulations.
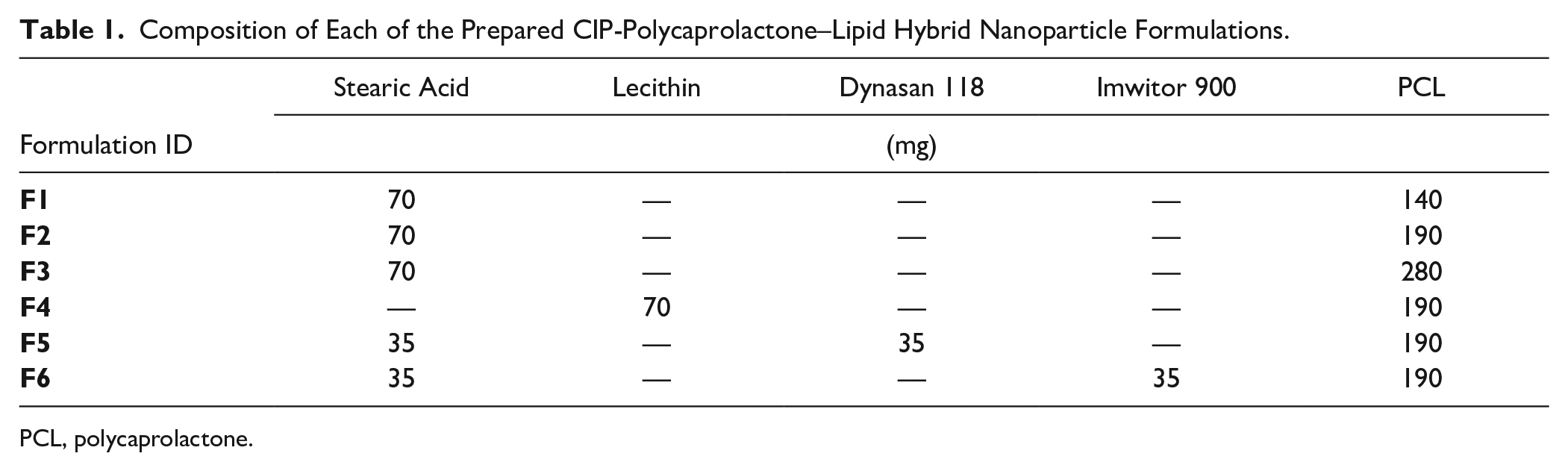
PCL, polycaprolactone.
Evaluation of the Prepared SLNs
Measurement of Particle Size and Zeta Potential
Samples from each formulation were dispersed in distilled water to give a concentration of about 0.1% and were measured for particle size and polydispersity index utilizing a ZetaPALS (Brookhaven Instruments Corporation, Holtsville, NY). The measurements were repeated three times using an angle of detection of 90°.
Measurement of Zeta Potential
The particle charge was obtained by determination of the zeta potential by applying the laser Doppler velocimetry (LDV) mode in the above Brookhaven instrument.
Measurement of Drug Entrapment Efficiency and Drug Loading
A sample of supernatant obtained after each centrifugation cycle in the preparation of each CIP-PLN formulation was filtered through a 0.45 µm membrane analyzed for the CIP using the above-mentioned spectrophotometric method. The percentage of drug entrapment efficiency (%EE) and percentage of drug loading (%DL) were determined according to the following formulas:
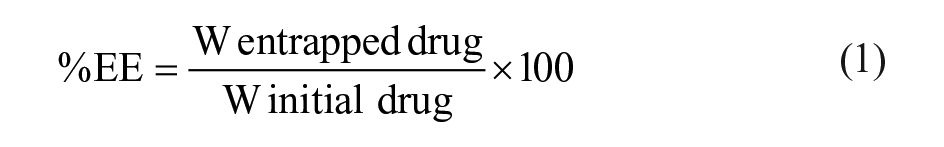
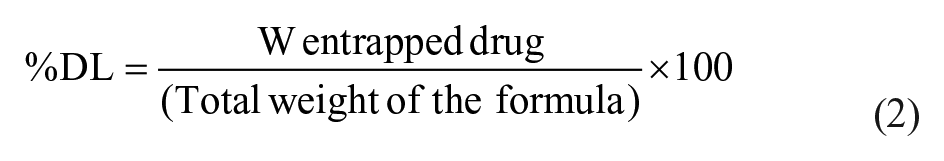
Particle Morphology
Scanning electron microscopy (JSM-6360 LV; JEOL, Tokyo, Japan) was employed for evaluation of the morphological and surface attributes of CIP-PLN formulations. The samples were gold coated in an argon atmosphere by applying a sputter module under a high vacuum evaporator (JFC-1100 fine coat ion sputter; JEOL) after prior fixing of the dried samples on carbon tapes. The photomicrographs were recorded at a 10 kV rate of acceleration.
Differential Scanning Calorimetry
Thermograms of CIP-PLN formulations and pure CIP were developed utilizing a Netzsch DSC 214-Polyma differential scanning calorimeter (Bavaria, Germany) after calibration with indium/zinc standards. The samples were examined over a temperature range from 20 to 300 °C with a 10 °C/min flow rate under nitrogen gas.
Fourier Transform Infrared Spectroscopy
Agilent Cary 630 FTIR (Agilent Technologies, Santa Clara, CA) was employed in this study. Briefly, 2–3 mg from pure CIP, drug-free (blank) PLN, or formulations F1, F2, and F4 were placed in the diffuse reflectance cell and exposed to the infrared light. The Fourier transform infrared (FTIR) spectra were determined from a 600 to 4500 cm−1 wavenumber range.
Drug Release Profile
The dialysis tubing method was used to monitor the CIP release profile from various PLN formulations. Certain weights from each formulation equivalent to 1 mg CIP were weighed and dispersed in 1 mL of phosphate buffer solution, pH 7.2. The dispersion is placed in a cellulose dialysis tubing, with a cutoff 12 KDa firmly tied from one end. After tying the other end, it was placed in a 25 mL flask containing 20 mL of the buffer. The closed flasks were then placed in a shaker water bath adjusted to 37 ± 1 °C and 80 rpm shaking rate. At certain time periods, 3 mL samples were taken from the outer medium and replaced with a preheated fresh medium. The filtered samples were analyzed spectrophotometrically for CIP concentration. Three samples from each formulation were tested and the means ± standard deviations of the cumulative percentage of CIP released were plotted against time for 6 days.
Antimicrobial Activity of CIP-PLN Formulations: Agar Diffusion Test
The antimicrobial activities of CIP-loaded PLN formulations F2, F4, F5, and F6 and pure CIP were tested against the clinical bacterial isolate (E. coli) EC04, obtained from the microbiology laboratory at King Abdulaziz Medical City Hospital, by a modified agar well diffusion method. Briefly, agar petri dish plates were prepared by pouring 25 mL of autoclaved Mueller Hinton Agar in each plate and were left until solidified. A hundred microliters of adjusted bacteria at 5 × 105 CFU/mL of 0.5 McFarland standard was added to each agar plate and spread evenly by a spreader. Each agar plate was divided into two halves to accommodate tested drug formulas separately for comparison. CIP-PLN formulations and pure CIP were separately added as twofold serial dilution on plates starting at 100 μg/mL (5 μL of each CIP-PLN formulation and CIP concentration was spotted on the plates correspondingly). Plates were incubated for 24 h at 37 °C, and the drug inhibition zone was measured (the diameter of the inhibition zone was measured in centimeters by digital microbiological calipers) to identify the antibacterial activity of each drug formula in comparison with corresponding concentrations of the other drugs.
Results
Particle Size and Zeta Potential
Figure 1 summarizes the results of particle sizes, polydispersity, and zeta potential for all CIP-PLN formulations. All the prepared PLN formulations showed low particle diameters in the nanorange, from 133.9 to 217.1 nm. The variation in particle size among the six PLN formulations reflects dependence on the lipid-to-polymer ratio. The highest size was observed with F1 containing a 1:2 stearic acid-to-PCL ratio. Reducing the ratio to almost 1:3 and 1:4 in F2 and F3, respectively, resulted in a significant reduction in particle size. The composition of the lipid counterpart also appeared critical for the size. F5, containing a lipid composition of 1:1 stearic acid to Dynasan, exhibited the lowest particle size value, while F6, containing a 1:1 composition of stearic acid to Imwitor 900, had a significantly higher value. The prepared particles showed reproducible particle sizes from the freeze-dried powder after dispersion in phosphate buffer, pH 7.2, for 20 min using a sonication bath.
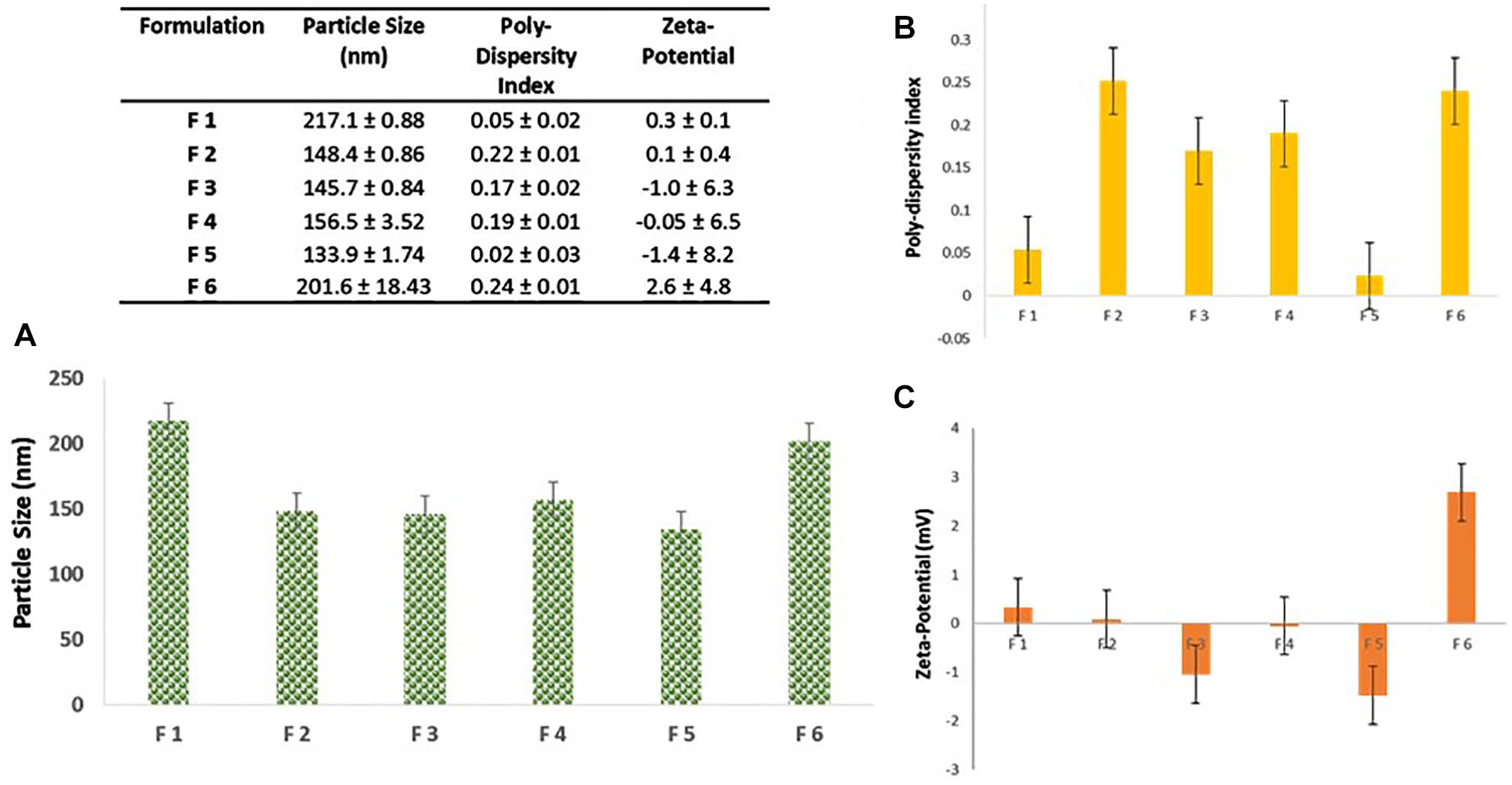
Physical characteristics of all PLN formulations. Bar charts of (
Generally, all the formulations showed low polydispersity index values ranging between 0.02 to 0.24, indicating a narrow particle size distribution range; it was consensually agreed upon in the literature that values below 0.3 are referred to as monodisperse. 28 This was also reflected from the low standard deviation values compared with the mean values of particle sizes. The lowest polydispersity value (0.03) was observed with F5, the formula with the lowest mean particle size.
The zeta potential results demonstrated in Figure 1 indicate low magnitude (below ±3 mV), with high variability indicated by the very high standard deviation values. The species of net charge varied from –Ve to +Ve. Thus, addition of a small amount of an electrolyte is recommended for stabilizing CIP-PLN when stored as a dispersion.
Particle Morphology
The particle morphology was inspected using scanning electron microscopy. The scanning electron microscopy images ( Fig. 2 ) indicated a uniform particle size in the same range indicated as that by light scattering measurement. All the images demonstrated the spherical nature of the prepared particles with smooth surfaces.

Scanning electron microscope images of different CIP-loaded PLN formulations: (
%EE and %DL
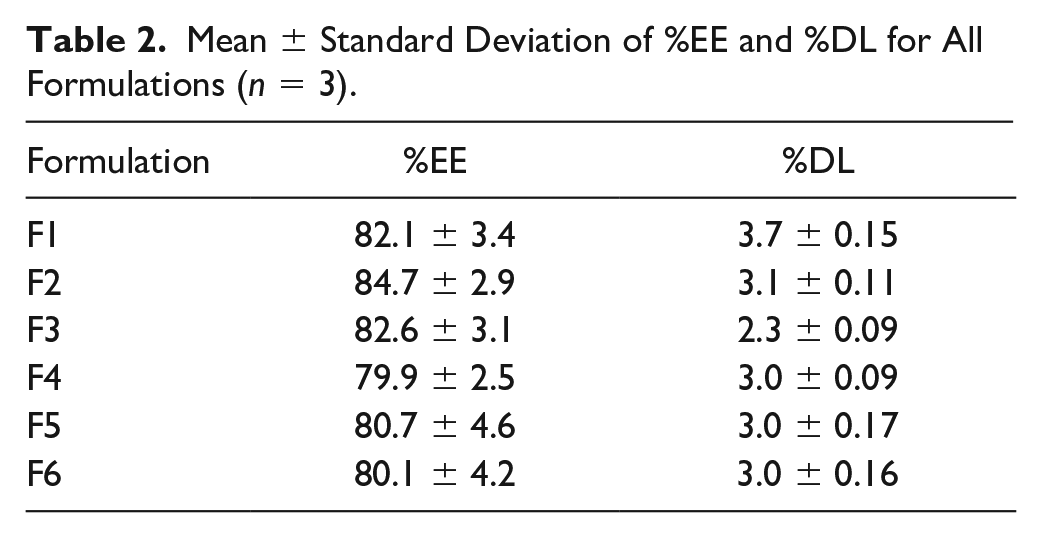
The values of CIP %EE in the six formulations were high, ranging from 79.9% to 84.7% (data are presented in Table 2 ). This proves the usefulness of the method of preparation in encapsulating large doses of CIP inside the particle matrix. Formulation factors such as lipid-to-polymer ratio and type of lipid material were not determinant factors for the efficiency of CIP entrapment inside the PLN formulation since there were no significant differences between %EE for all six formulations. All the formulations showed low %DL values ranging from 2.3% (F3) to 3.7% (F1). The use of low drug-to-PCL ratios, from 1:14 to 1:28, in addition to the incorporation of 70 mg of lipid in each formula, led to such low %DL values. However, the total weight of the dose can tolerate the use of multiple folds of such values as per the various therapeutic needs.
Mean ± Standard Deviation of %EE and %DL for All Formulations (n = 3).
Thermal Behavior
Figure 3 depicts the thermal profiles of all the prepared formulations in comparison with pure CIP. The thermogram of pure CIP shows a sharp endothermic peak at 272.3 °C corresponding to its melting. This is very close to what was reported earlier for a CIP melting peak at 268.0 °C. 27 All the formulations’ thermograms showed endothermic peaks at low temperatures below 80 °C corresponding to the melting of PCL and lipid components of the formula. The shifting of one or all peaks was demonstrated in some formulations, indicating possible interaction between PCL and the lipids.
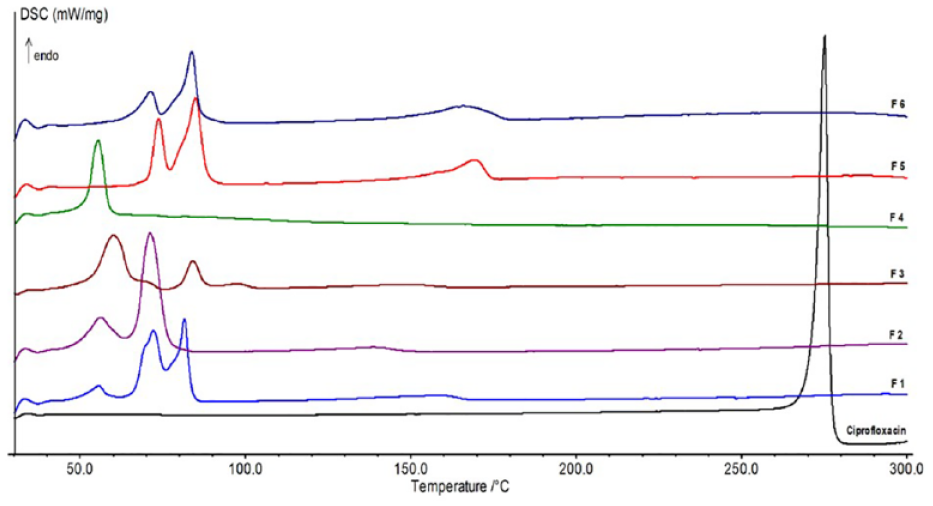
DSC thermograms of pure CIP and CIP-loaded PLN formulations.
FTIR
Figure 4 depicts the FTIR spectra of pure CIP, blank PLN, and F1, F2, and F4. The spectrum of pure CIP showed a medium band at around 1020 cm–1 wavenumber corresponding to C–F stretching, a strong band at 1270 cm–1 corresponding to bending of the hydroxyl group, a band at 1392 cm–1 due to asymmetric stretching of the carbonyl group, a strong band at 1490 cm–1 indicative of the stretching vibration of O–C–O of carboxylic, and a strong band around 1600 cm–1 related to the N–H bending vibration of the quinolone ring. The blank PLN spectrum reflected the strong characteristic bands of both PCL and stearic acid, including the strong bands at 2890 and 3050 cm–1 corresponding to symmetric and asymmetric CH2 stretching, respectively; a strong band at 1725 cm–1 indicative for carbonyl stretching; and a strong band at 1165 cm–1 indicative of both C–O and C–C stretching in the amorphous phase. The spectra of F1, F2, and F4 expressed the main characteristic of the blank PLN and broadening and slight shifting of the main characteristic bands of CIP.
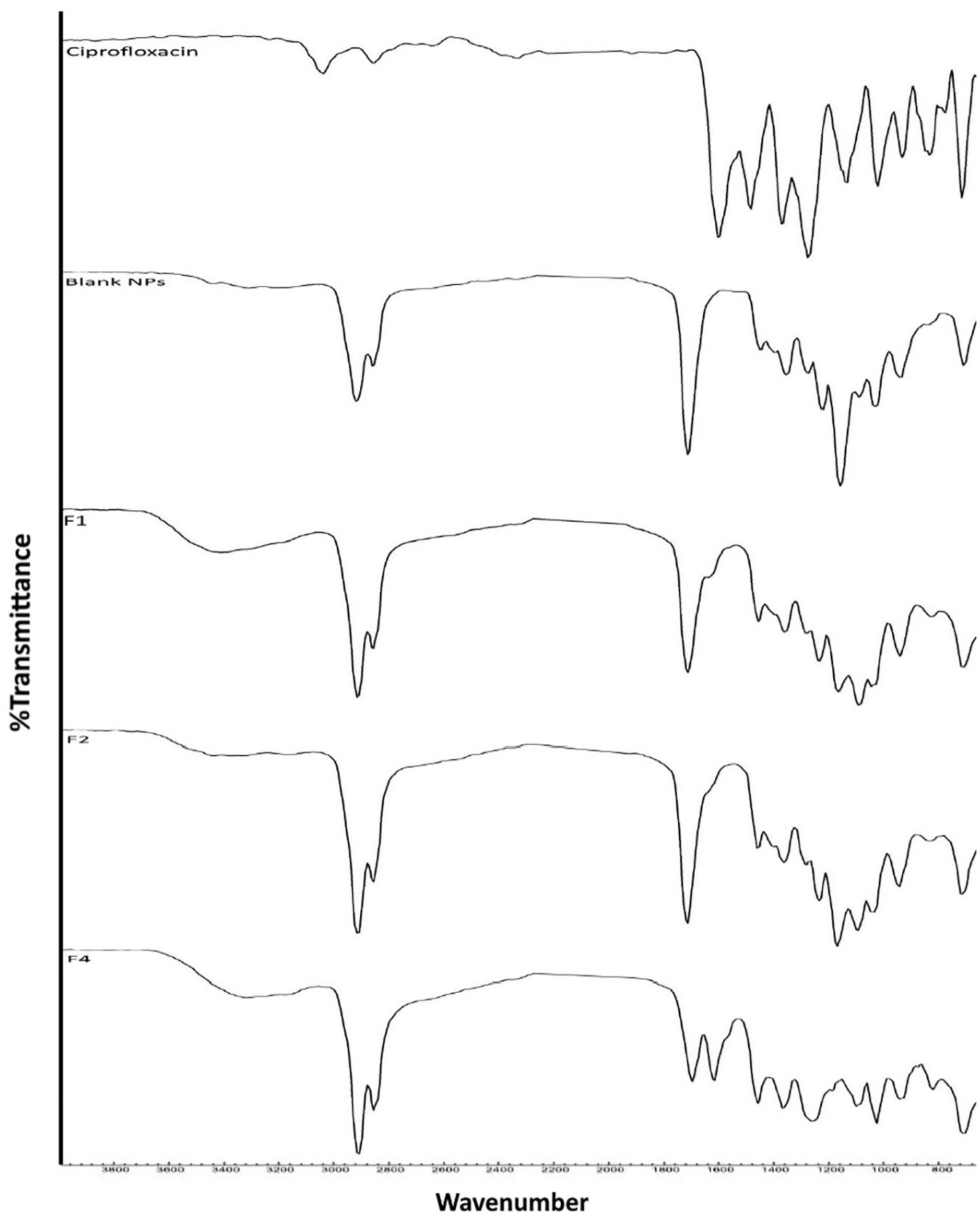
FTIR spectra of pure CIP, blank PLN, and CIP-loaded PLN formulations.
CIP Release Profiles
The CIP release profile from the six CIP-PLN formulations was monitored for 6 days and the results are depicted in Figure 5 . Generally, all the formulations exhibited a slow release profile with highly variable rates and no burst release. F5 and F6, containing a combination of stearic acid with either Dynasan or Imwitor in a 1:1 ratio, exhibited a significantly faster CIP release rate than the other formulations, including F2, containing the same ratio of PCL. This shows that the enhancement in the CIP release rate is directly linked to the inclusion of the triglycerides (Dynasan and Imwitor) in the formula. Comparing the release profiles of F1, F2, and F3, it is conclusive that the lower stearic acid-to-PCL ratio is accompanied by a faster CIP release rate. The use of lecithin as the only lipid resulted in a significantly lower release rate compared with all CIP-PLN formulations, as shown from F4 profile. It exhibited only a 20% cumulative release after 3 days and reached ≈30% after 6 days.
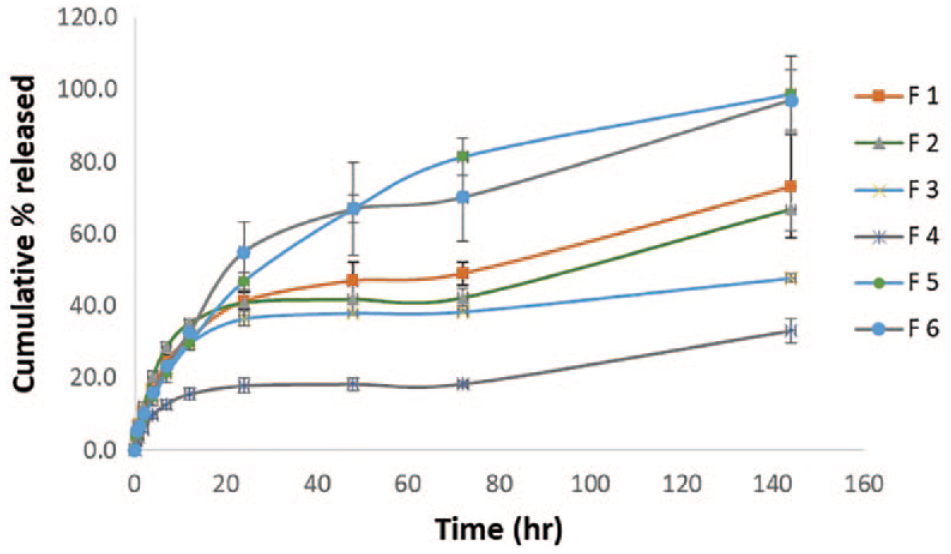
CIP release profile from six PLN formulations. Antibacterial activity comparison between CIP-loaded PLN formulation F4 and pure CIP.
Antimicrobial Effect Studies
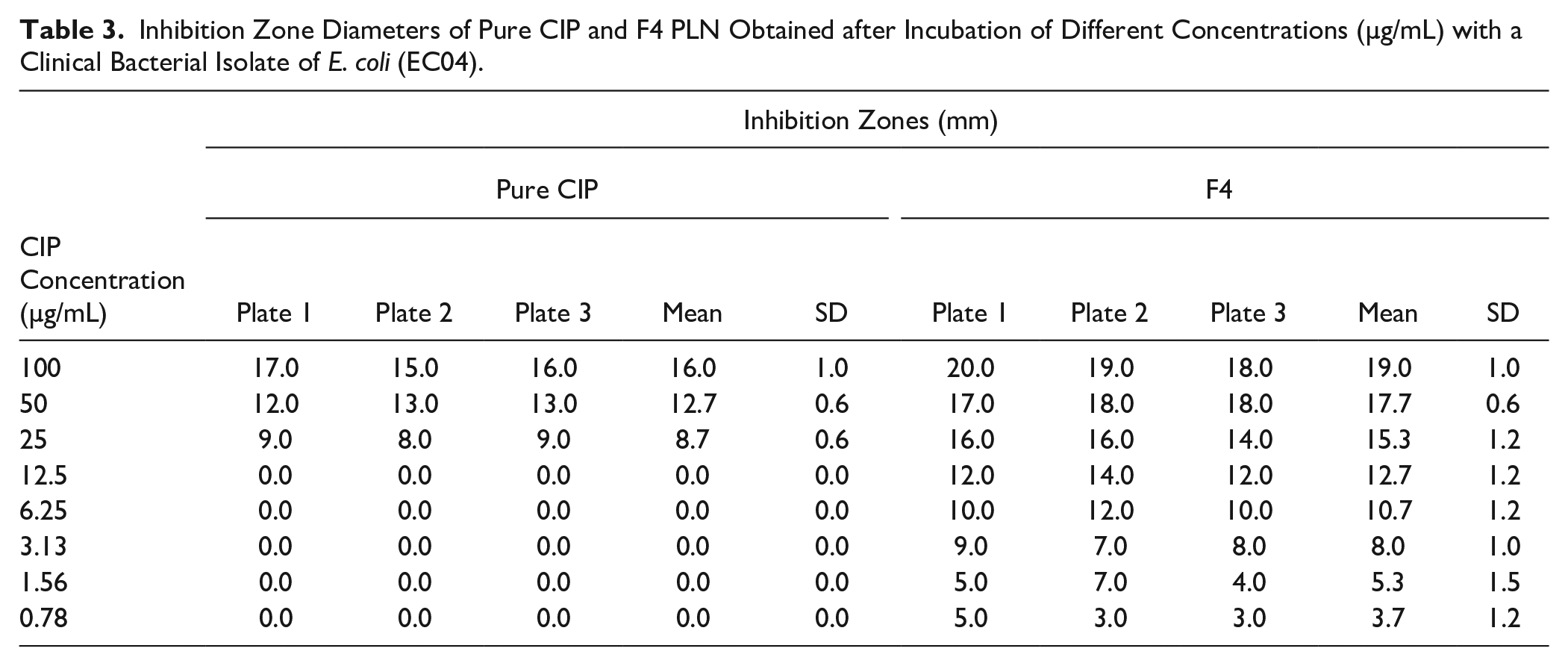
The clinical bacterial isolate E. coli (EC04) was used to examine the antibacterial activity of the CIP loaded in PLN formulations. Among the four tested formulations, only F4 illustrated significant antibacterial activity in comparison with the free CIP formula. Table 3 presents the measured diameters of zones of inhibition (mm) of F4 formulation compared with pure CIP after incubation of 100, 50, 25, 12.5, 6.25, 3.125, 1.56, and 0.78 µg/mL equivalent CIP concentrations. F4 inhibited EC04 growth at concentrations as low as 0.78 µg/mL, while pure CIP failed to inhibit the growth when diluted below 25 µg/mL. There was a significant increase in the diameters of the inhibition zones of F4 compared with pure CIP for 100, 50, and 25 µg/mL plates.
Inhibition Zone Diameters of Pure CIP and F4 PLN Obtained after Incubation of Different Concentrations (µg/mL) with a Clinical Bacterial Isolate of E. coli (EC04).
Discussion
In this study, a new single-step method for PLN preparation was introduced with novel composition to enhance the delivery of CIP through prolonging drug release and fostering the bioavailability and antibacterial activity. The produced PLN formulations exhibited the attractive physical characteristics of uniform low particle sizes and regular spherical shapes. PCL was chosen as the skeleton of the particle matrix because of its superior physical and mechanical attributes over many other biodegradable polymers.15,29 In addition, PCL has unique rheological and biological properties.30,31 This explains its widespread applications in drug delivery and implantable therapeutic systems.32,33 The PCL polymer chain imparts high flexibility in producing uniform low nanoparticle sizes, as observed with CIP-PLN. It was reported that compared with PLGA, PCL is capable of producing lower nanoparticle sizes.34,35 The lipid-to-polymer ratio was adjusted to a minimum of 25% (F3), as it was reported by Chan et al. 36 that increasing the lecithin-to-PLGA ratio from 10% to 20% resulted in significant reduction in PLN particle sizes. Increasing the ratio up to 50% (F1) did not affect the particle size. Varying the lipid composition from a single element (F1–F4) to two elements (F5, F6) was not found to be a determinant of either particle size or polydispersity indices. This is in contradiction with what was recommended by Troutier et al., 37 that the use of a single lipid is necessary to achieve narrow particle size distribution.
All the formulations exhibited favorable a %EE around 80%, which is similar to what was reported by Mieszawska et al. 38 In addition, the lipid-to-polymer ratio was assumed to be a critical parameter for enhancing %EE, as reported by other studies that illustrated a maximum %EE for two anticancer drugs in lipid–polymer nanoparticle formulations using lipid-to-polymer ratios of 1:5 and 1:7.15,36 Controversially, our study illustrated the independent effect of %EE on the lipid-to-polymer ratio since no significant difference in %EE between F1 (ratio 1:2) and F3 (ratio 1:4) was recorded. This can be attributed to the similarity in the core composition of all PLN formulations (PCL) and the use of a common solvent (chloroform), which allowed for deep impedance of CIP into the PCL matrices.
The disappearance of the melting peak of CIP from the differential scanning calorimetry (DSC) thermograms of all formulations indicates the existence of CIP in an amorphous state. Based on consensus interpretation of many similar results in the literature, this phenomenon can be counted for the possibility of complete solubilization of CIP within the PLN matrix.39–41 This was also concluded from the slight shifting of the characteristic CIP bands in the FTIR spectra of PLN formulations, indicating deep incorporation of molecular CIP within the polymer matrix. The disappearance of the lecithin melting peaks from F4 thermograms indicates the loss of its characteristic crystalline structure reported by Dora et al. 42 The existence of lecithin in an amorphous state hinders high flexibility, allowing for the formation of a phospholipid bilayer shell on the surface of the PCL core. This may explain the extreme delay in CIP release from F4. This agrees with the work of Chan et al., 36 who demonstrated that lecithin can self-assemble on the surface of PLGA nanoparticles, forming a liposomal bilayer of phospholipids.
The significant delay in release observed as a result of increasing the ratio of stearic acid to PCL can be attributed to the increase in the thickness of the lipid shell. The absence of the burst release in all PLN formulations was postulated as an approach to improve SLN outcomes. In the presented formulation, CIP is incorporated deep in the core PCL matrix, while the lipid shell layer, free of CIP, can induce a hindrance to the water influx into the polymer core and act as a release controlling layer. 43
The insignificant antibacterial activity observed with F2, F5, and F6 after incubation for 24 h can be expected from their slow CIP release rates, less than 60% after 24 h. Thus, they may cause enhancement in the antibacterial activity in vivo. This was also noticed by Jeong et al., 25 who reported higher in vivo antibacterial activity of CIP-PLGA nanoparticles in mice despite the observed lower in vitro antibacterial activity compared with pure CIP. They attributed this observation to the extreme prolongation in drug release induce by PLGA nanoparticles. 25 The extreme potentiation of the antibacterial activity against EC04 achieved with F4 is highly attributed to the liposome-like shell formed by lecithin, leading to higher cellular uptake. 44 This is in agreement with Zhang et al., 45 who reported that lecithin significantly enhanced the antibacterial activity of eugenol against three strains of E. coli. The highly significant antibacterial activity effects of F4 has been achieved despite the extremely low percentage (≈20%) of CIP released in 24 h (incubation period) through the possible internalizing of CIP inside the bacterial cell. It is expected that this effect can be highly magnified in vivo, leading to significant reduction of the CIP doses and thus significant reduction in all its dose-dependent toxicities. The ratio of lecithin and/or PCL can be optimized toward faster release options to allow for more clinical applications.
The proposed PLN systems showed unique superior attributes in enhancing the delivery of CIP. The incorporation of PCL allowed for higher physical stability and well-controlled uniform CIP release prolongation, while the incorporation of lipids provided higher cellular uptake and bioavailability and biocompatibility. The incorporation of lecithin showed very interesting outcomes in enhancing the antibacterial activity. The overall therapeutic efficacy of CIP would be enhanced through reduction of the therapeutic doses and, consequently, all dose-dependent adverse effects. This system has the advantage of being able to be utilized for broad clinical application through many routes of administration, including parenteral and implantation, oral, pulmonary, and ophthalmic.
Conclusion
We reported a novel single-step method for the self-assembly of CIP-PLN systems. The prepared CIP-PLN formulas showed low uniform nanoparticle size, high CIP %EE, and prolonged release rates. Interestingly, F4, containing lecithin, significantly enhanced the antibacterial activity of CIP. This can lead to significant improvement of the therapeutic efficacy of CIP. Further studies are recommended to pursue these beneficial attributes toward useful clinical applications.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from King Abdullah International Research Center, National Guard Health Affairs, Riyadh, Saudi Arabia (grant no. SP17/391/R). The funding agency had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.