
Abstract
The discovery of new medicines has become increasingly more challenging and requires significant collaboration between pharma, biotech, academia, and technology to be successful. These partnerships necessitate the streamlined exchange of samples while adhering to the increasingly complex set of legal and proprietary restrictions, government legislation, and ethical considerations associated with samples. There is a significant volume of literature published on clinical sample compliance but little describing compliance aspects of discovery sample management. This paper describes some of the key compliance activities and challenges and shares GlaxoSmithKline’s experiences and current practices.
Introduction
In the last 15–20 years, sample management has evolved highly sophisticated processes and operations to support drug discovery, 1 overcoming the automation challenges to provide solid and solution small-molecule libraries at scale. 2 Quality assurance associated with the process and the samples was addressed by the application of modern analytical platforms and by the adoption of Lean Sigma practices.3,4 In recent years, the scientific community has begun to recognize the synergies between the tools, practices, and skill sets associated with small-molecule and biological sample management, and groups that traditionally focused on compounds are now broadening their remit to cover other sample types. The different disciplines that cover the broad elements of sample management, for example, small molecule, biologic, and clinical samples, are recognizing the synergies between these areas and are coming together as a community to share best practice and learnings.
Parallel to these technology developments, the drug discovery paradigm has become a truly collaborative enterprise where most organizations operate with multiple alliances, collaborations, and external contracts. 5 With this external focus, it becomes increasingly important to have robust compliance processes in place. Compliance management is the process by which managers plan, organize, control, and lead activities that ensure compliance with regulations, laws, and standards. Each organization needs to assess its own level of risk exposure, but in general the cost of noncompliance far outweighs the cost of prevention ( Fig. 1 ). Compliance aspects associated with sample management are becoming more critical as new legislation is developed and as research becomes more collaborative and distributed across sites and countries. Organizations need the ability to track and trace what materials and data they have internally and externally. They need to know what can be transferred where, move samples across borders efficiently, ensure timely delivery, guarantee compliance with any restrictions, and protect intellectual property (IP). Having robust compliance processes in place is a fundamental component of an organization’s license to operate and is part of its core values.
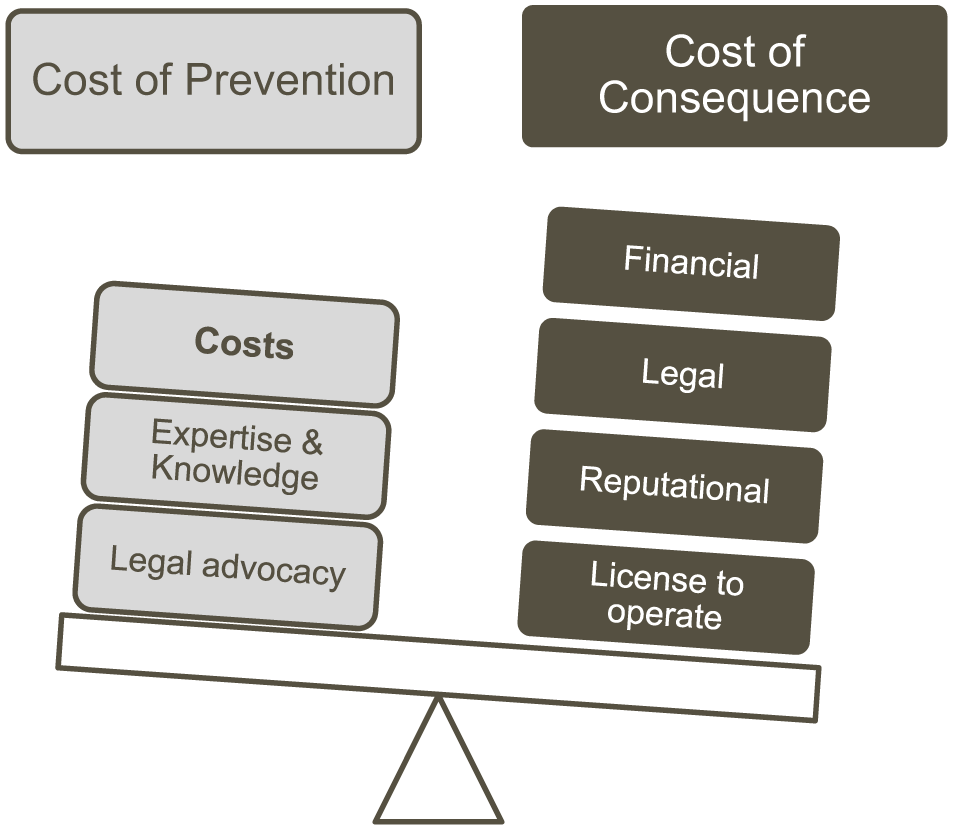
The risk equation. Balancing the cost of prevention versus the cost of consequences.
To date, little has been published on compliance within the nonclinical sample management discipline. The focus of this article is to provide a review of best practice and guidelines covering compliance approaches to material transfer; the management of assets and treatment of controlled drug strategies; risk mitigation for biological sample use, including licenses and the Nagoya Protocol (NP); and shipping.
Material Transfer Agreements
In moving toward collaborative research,6–8 most research staff lack the legal compliance authority and knowledge to enter into a contract. 9 With half of all drugs today being developed through collaborations, 10 and many of these requiring the movement and transfer of samples, it has become critical that sample management organizations develop expertise and a working knowledge of agreements to provide materially externally.
A material transfer agreement (MTA) is a legally enforceable contract employed by research institutions and companies to set the terms under which their materials and associated data may be obtained and used by others with no funding involved11,12—a place where law, science, and goodwill collide. These agreements provide a mechanism to protect the interests of the owners of discoveries and inventions, while promoting data and material sharing in the research community. 5
Agreement specialists must adhere to various levels of internal and external compliance obligations while producing a legally binding document to facilitate the transfer of materials. Prior to drafting an agreement, the specialist has several internal compliance checks to undergo. Determination of whether the material is an asset or part of an ongoing alliance agreement is key, as this will dictate what level of IP protection is required. If the material being transferred is part of an alliance or consortium, the governing agreement (e.g., license or research collaboration agreement) needs to be consulted to ensure that the material can indeed be shared externally and to determine if there are any provisions that need to be included in any subsequent agreements. Should the material be of candidate quality, further internal approvals are generally required. For both candidate and alliance material, the specialist must ensure that all appropriate approvals from project teams have been obtained and documented. Additional checks should be made to ensure that the material is available in inventory, meets quality standards 13 for provision externally, and is not regulated in the recipient’s country. 14 It is also necessary to review the proposed research checking for any potential animal or human biological sample (HBS) usage. All animal and HBS usage require separate compliance procedures to guarantee that both the provider and recipient adhere to all governmental regulations.15,16 Lastly, to comply with record retention policies, an official record of the agreement will be generated in the organization’s legal databases.
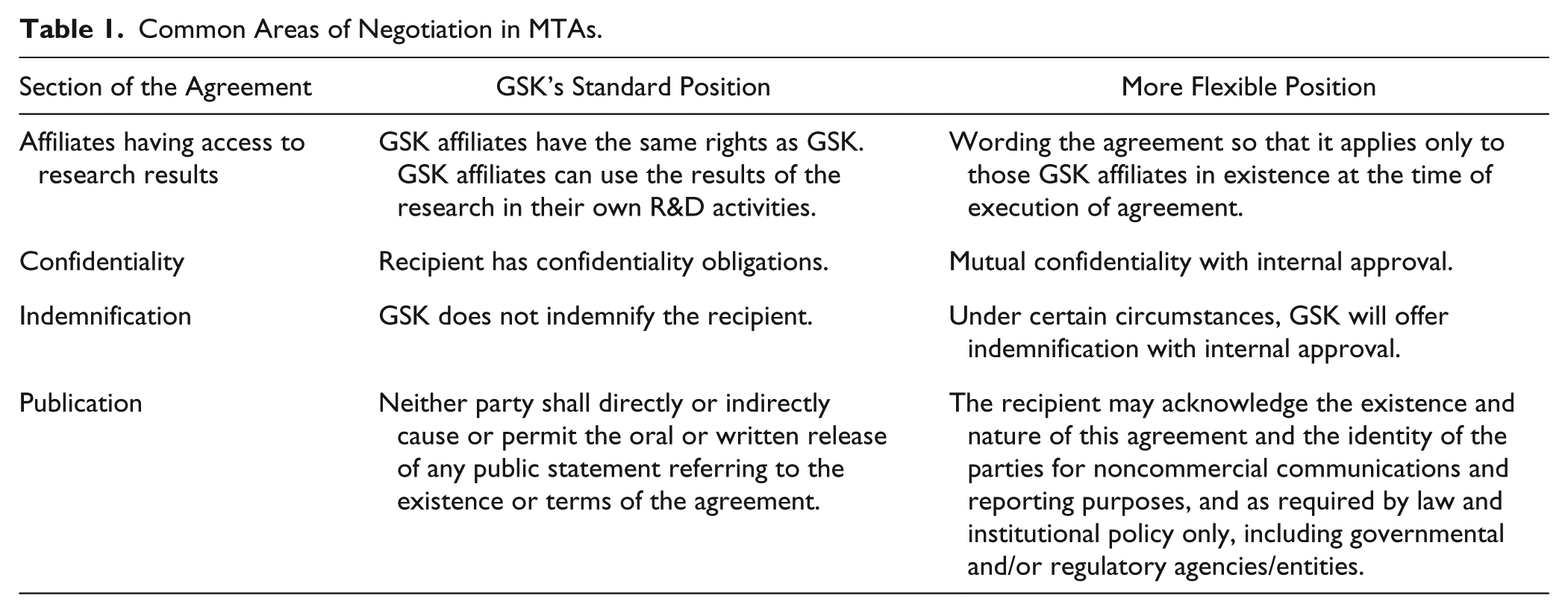
After addressing internal compliance requirements, a first draft of an outbound MTA is generated and sent to the recipient institution. The draft agreement usually goes through several iterations before it is signed. This is often due to publication, indemnification, and IP concerns, which can typically be overcome with some flexibility ( Table 1 ). Negotiators are trying to protect IP, while adhering to internal policies and promoting a scientific community that shares materials and data. 10 IP issues regularly arise when government funding is used for the research of the MTA. The Bayh-Dole Act 17 allows academia the ability to elect ownership of inventions made during research that utilizes federal funding and to become directly involved in the commercialization process.18,19 Industry typically pushes back on Bayh-Dole unless the specific research associated with the MTA utilizes government funds. In cases of dispute, the terms of an MTA can play a role in determining the ownership of patents and the assignment of IP rights.10,12 Other areas of regular discord typically include publication, liability, and indemnity.16,19
Common Areas of Negotiation in MTAs.
MTAs have a defined finite term, and therefore the incorporation of agreement specialists within the sample management line facilitates the rapid transfer of material to the external party. The combination of specialist legal knowledge and expertise in the sample management operation, including thr ordering and shipping of materials, is a powerful enabler of partnerships and collaborative research.
Control of Small-Molecule Assets: Post–Candidate Selection
A candidate drug is a molecule (small molecule, antibody, etc.) with strong therapeutic potential and whose activity and specificity have been optimized. Typically, from target identification to candidate selection can take 1–3 years and represents a significant investment for an organization. The control and release of assets post–candidate selection must therefore be carefully managed for exploratory work within discovery or non-GxP (Good Practice Quality Guidelines and Regulations) areas. The project leader or manager needs to approve the use of the post-candidate-selected assets for nonclinical and non-GxP studies as well as have visibility of GxP work, and the outcomes of these studies. This is so that any potential safety signals can be detected to allow the evaluation of the overall risk profile of a molecule. Tracking of all releases of a compound allows the project leader to be aware of all results and then to ensure that that the appropriate data are included in the investigator brochure after consultation with regulatory groups within their organization. Management monitoring on dispensements from compound inventories should be checked against tracking tables to ensure that full disclosure of all work undertaken has been recorded.
The full asset ID (compound number) and batch must be clearly cited in all documentation to ensure that full data integrity is maintained and tracked. Abbreviations and/or local nomenclature should always be avoided. Tagging to the lot and batch level is required, as this then allows for the detection of any specific safety implications with a batch or lot number.
The release of postcandidate asset needs to be controlled not only during the life span of active projects associated with the asset, but also across release to any project. Projects using a nonactive asset (an asset that is not associated with any ongoing research in-house) also need to be aware of their responsibilities in ensuring that any adverse results are reported and that data can be retrieved as and when required. The specific responsibility of sample management groups is to ensure that postcandidate assets are appropriately tagged and that requests for access to such compounds are held until approved by the project leader and the approval is tracked.
Regulated Compounds in the Pharma Industry
The global nature of scientific research, for example, in the pharmaceutical business, contract research organizations, and academia, requires that these organizations have knowledge of the worldwide legislation governing small molecules and biological materials. This is to ensure compliance with legislations, not only when transporting materials within an organization across global sites but also when transferring to an external group. The scheduling of clinical trials and manufacturing also needs careful consideration to ensure that country rules are not violated.
Some prescription medicines are controlled under legislation for the misuse of drugs (and subsequent amendments). These medicines are called controlled medicines or controlled drugs. Pharmaceutical companies routinely work with controlled substances and must have adequate controls in place to meet the legislative requirements of the countries they operate in.
Controlled drug legislation is constantly evolving and becoming more complicated as it attempts to mirror developments in substance abuse. The burden of regulations that need to be adhered to is complex and extensive, and Table 2 lists just a selection of current worldwide regulations. There is no sole source of worldwide legislation to refer to for pharma industries.
List of Legislation Covering Controlled Drugs.

Difficulties are increased by the fact that legislative bodies can sit at an international, area, country, or state level, which adds to the challenges of ensuring adherence. For example, in the United States, the use and possession of cannabis is illegal under federal law for any purpose, by way of the Controlled Substances Act of 1970 (CSA). 20 Under the CSA, cannabis is classified as a Schedule I substance, determined to have a high potential for abuse and no accepted medical use. However, at the state level, policies regarding the medical and recreational use of cannabis vary greatly, and in many states conflict significantly with federal law. 21
The risks to an organization of noncompliance with controlled substance regulations are significant, in terms of not only fines and revocation of licenses, but also loss of reputation. There are many well-documented examples where organizations have been fined significant sums by the U.S. Drug Enforcement Administration for failing to comply with the CSA. The penalties for nonadherence to legislation are high and affect both pharma companies and distributors, as can be seen by the latest fines, stemming from the opioid crisis in the United States, for McKesson Corp., where they agreed in January 2017 to pay a $150 million fine. 22 Another example is Cardinal Health, who reached a $44 million settlement. 23
UK Schedule 1 and 2 drugs are heavily regulated and include drugs such as morphine and 3,4-dimethoxybenzeneethanamine. The use of controlled substances is approved for individual researchers and only for the research location(s) described in their Drug Enforcement Administration application. For sample management groups, all samples must be registered, barcoded, and stored in government-approved lockable cabinets, which must be securely bolted to a wall. Only limited personal with appropriate training should have access to such stores, and material can only be distributed to licensed authorized scientists. In principle, if required, larger quantities of controlled chemicals could be stored in a secure, lockable room, which may require intruder alarms and CCTV. In addition to data captured by an inventory system, all transactions must be recorded in hard-copy notebooks or registers including disposals. Controlled drug licenses are held centrally within an organization and allow the use of any compounds listed under these schedules.
Controlled Substance Expert Group Part of the Pistoia Alliance
The Pistoia Alliance is a global, not-for-profit alliance of life science companies, vendors, publishers, and academic groups that work together to lower barriers to innovation in R&D. The Controlled Substance Expert Group was formed out of an unmet need to define an application that would allow organizations to check their compound libraries against worldwide drug legislation in an automated and efficient manner. 14 This group has grown into an established cross-industry body, comprising representatives from global pharma and commercial enterprises. The breadth of this group’s expertise enables it to explore and compare the differing legislations worldwide, with an interest on how de-minimis limits and R&D clauses are framed. The goal is to achieve global parity on de-minimis limits by working with the International Narcotics Control Board (https://www.incb.org/).
Currently, the Controlled Substance Expert Group is setting up working groups with national governments. For example, in the United Kingdom, the group is working with the Advisory Council on Misuse of Drugs (ACMD) on how R&D exemption clauses could be applied within the United Kingdom. This is a critical issue, as current exempt product and de-minimis limits for Schedule 1 compounds indicate that no one component part of the product or preparation can contain more than 1 mg of the controlled drug (or 1 µg in the case of lysergide or any other N-alkyl derivative of lysergamide).
With these de-minimis limits, it is very difficult to carry out late discovery or development on Schedule 1 compounds in a global arena without having the appropriate letter of no objection and export licenses. In addition, the consequences of new generic legislation (see the following case study) means that more compounds within organization screening decks come under these restrictions, preventing the shipment of these collections across organizations and countries.
Legislation Wording Changes: Third-Generation Synthetic Cannabinoids—A Case Study
Historically, compound names were specified in controlled substance legislation, which required translation into scientific nomenclature. This was relatively straightforward for companies to interpret. Now, there is an increase in the use of generic structures, as exemplified in the Misuse of Drugs Act 1971 Amendment Order 2016. 24 The act addresses third-generation synthetic cannabinoids and promotes a completely new type of generic definition ( Fig. 2 ).
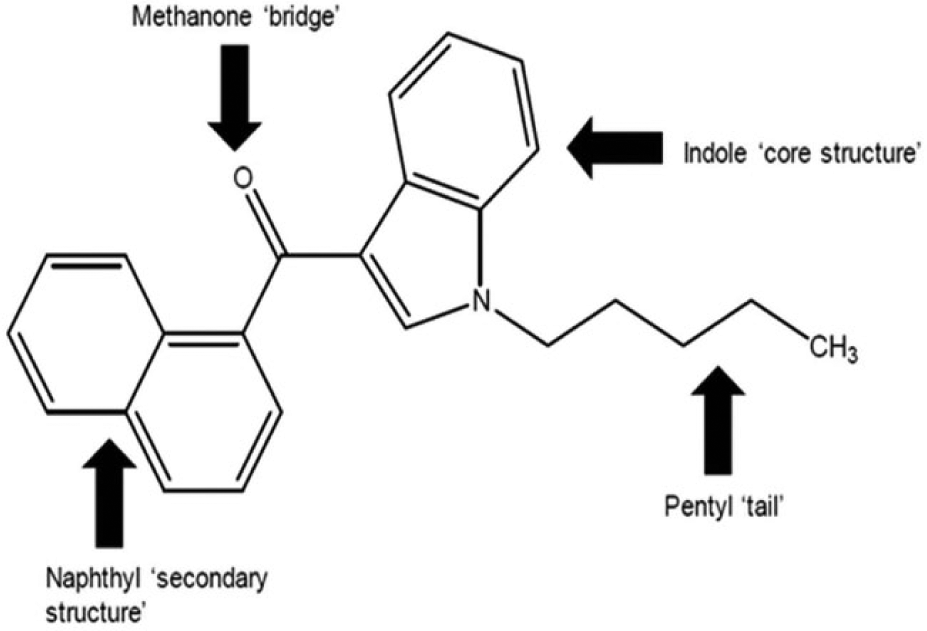
The act gives examples of the modifications that can be made at any of the four substructures indicated that will also place the resulting structure into Schedule 1.
The problem with such generic definitions is that they capture some already valuable commercially available medicines, and consequently, these needed to be exempted from the Misuse of Drugs Act 1971 Amendment Order 2016. Not only are these proven medicines, and so do not fall under Schedule 1, which is specifically for substances considered by the government to have no medicinal value, but also they do not exhibit any cannabinoid-like activity (CB1). Examples of these include atorvastatin (statin), losartan (angiotensin II receptor antagonist for hypertension), and zafirlukast (antiasthmatic).
The impact of this amendment is that typical pharmaceutical libraries have seen their controlled compounds rise into the tens of thousands, with most having no indication of CB1 activity. This has prevented hit identification and lead optimization activities and limited collaborative research due to the inability to ship these compounds outside the United Kingdom. Of even more concern is the impact on the ongoing supply of investigational medicine for several pharma companies. To avoid an interruption to their dosing regimen to patients, GlaxoSmithKline (GSK) had to work closely with two separate clinics, supporting them in their application for the appropriate home office licenses to ensure that these were in place before the next dispensing date. This date had to be rescheduled to allow for this additional activity. In parallel to this, all existing stocks of the investigational drug, whether held at a GSK site or locally at the clinics, required additional labeling and re-release by a qualified person. GSK staff traveled to the clinical sites to relabel locally held stocks. Future UK-based studies for this compound will be restricted to clinical sites that either already hold the appropriate home office license or are willing to meet the criteria to gain such a license. Movement of clinical materials into and around the United Kingdom places additional burden on GSK and its authorized shippers due to the need for additional permits and licenses. On this occasion, there was no impact on the patients or interruption to their supply of the investigational drug, but the additional constraints imposed by the Misuse of Drugs Act 1971 Amendment Order 2016 place an added burden on the clinical supply chain to ensure that this remains the case.
This legislation is now difficult to interpret without the aid of commercial controlled substance identification packages, for example, Controlled Substance Compliance Checker by Scitegrity and Compliance Checker by Chemaxon. This is due to the sheer number of possible structures resulting from the permutations of the generic substituents. Academic laboratories and smaller biotech companies without access to these software packages are virtually blind to any Schedule 1 compounds within their libraries due to the complexity of interpreting the Markush structures. This potentially leads to inadvertent use of the compounds outside of a licensed premise, as well violating import and export regulations.
Because of the sheer complexity of the legislation, it is beholden to the pharma industry and partners to self-declare compounds falling under this legislation. It is not possible for licensing bodies to scrutinize shipments unless full structures are available for checking against either of the two noted software packages. Accordingly, GSK has implemented an automated check of all materials prior to shipment.
There needs to be better liaison between regulators and pharma advocacy channels to ensure that genuine research is not hindered. This needs to be through earlier conversations around the scope of generic clauses and de-minimis limits. The scope of the new legislation should be determined by testing on commercial and pharma libraries to gauge effect. Would the compounds captured by the legislation exhibit the undesired effect and are the numbers proportionate? Considerable progress is being made to ensure that increased consultation and advocacy take place to ensure that medicine discovery is supported while maintaining appropriate controlled drug guidelines. The Pistoia Alliance is a good example of such a cross-industry advocacy group,
Biological Material Compliance
Until 10 years ago, the bulk of new medicines came from synthetic small-molecule compounds; however, by 2009 60% of all patent filings from big pharma (top 10 companies) covered biologicals. 25 This, coupled with the explosion of genetic data enabling increased interrogation of the biology related to disease states, has seen an increased uptake and use of biological materials within organizations. What today we consider sample management originated from the small-molecule operations as pharmaceutical organizations started to build chemical libraries for screening. 1 Although many of the tools and practices supporting small molecules have been leveraged to support biological sample management, there are unique challenges for biological sample management, such as automation at lower temperatures. The compliance issues for biologicals can also be very different, and emerging legislation such as Nagoya and biolicensing adds to that complexity. This section describes some approaches to meeting these compliance challenges.
Importance of Biolicensing
Protecting IP is well known to be of utmost importance in drug discovery, with robust compliance measures in place and enforced by legal and compliance professionals, but often less thought has been given to acquired third-party IP. Third-party IP is involved in sample management practices when biological materials and technologies are acquired from many sources, including commercial suppliers, academic institutions, government organizations, and collaborators, and is a common occurrence in modern drug discovery. The permitted uses of said biological materials and technologies are set forth by the terms of the licenses under which they were acquired, some of which are biological material licenses, limited use label licenses (LULLs), technology transfer licenses, patent and/or know-how licenses, and MTAs. Mostly these licenses protect patents, a temporary grant of a monopoly on the right to make, use, offer for sale, or import an invention in a country where the patent is in force. 26
Universities and federal laboratories can claim ownership and receive financial benefits from licensing their inventions through the negotiation of MTAs or licensing agreements, 27 which include specific terms and conditions on the permitted uses. Life sciences companies have routinely and customarily placed limitations on the postsale use of patented items. 28 These restrictions are often outlined in label licenses, which are commonly used in the life sciences industry to, for example, limit the use of a product to research and not diagnostic purposes, restrict resale, or ensure that no implied license extends from, for example, an instrument to reagents. 28
In 1996, the NIH established a license monitoring and enforcement group to supplement the traditional patenting, marketing, and licensing activities and, as a result, has collected about $17 million USD in unpaid and underpaid license royalties through formal financial audits and other investigative activities. 29 An example of breach of license terms is the case of a researcher at the University of Pittsburgh being criminally prosecuted for mail fraud for ordering microbial materials from the American Type Culture Collection using his institutional approved account on behalf of a fellow researcher at an unapproved institution, in violation of an MTA that prohibited transfer of materials to third parties. 9
Therefore, obtaining the appropriate licenses, understanding their terms, and tracking their use, as well as doing the same for the biological materials and use of technology associated with them, is critical to avoiding financial, legal, and reputational damage from patent infringement or breach of the license. GSK has recently implemented more automated measures to ensure compliance with licenses through a mandate to register biological materials and by the development of a centralized system to store and track licenses and their terms. This will enable restriction of use of the biological samples according to license terms by linking these IT systems ( Fig. 3 ).
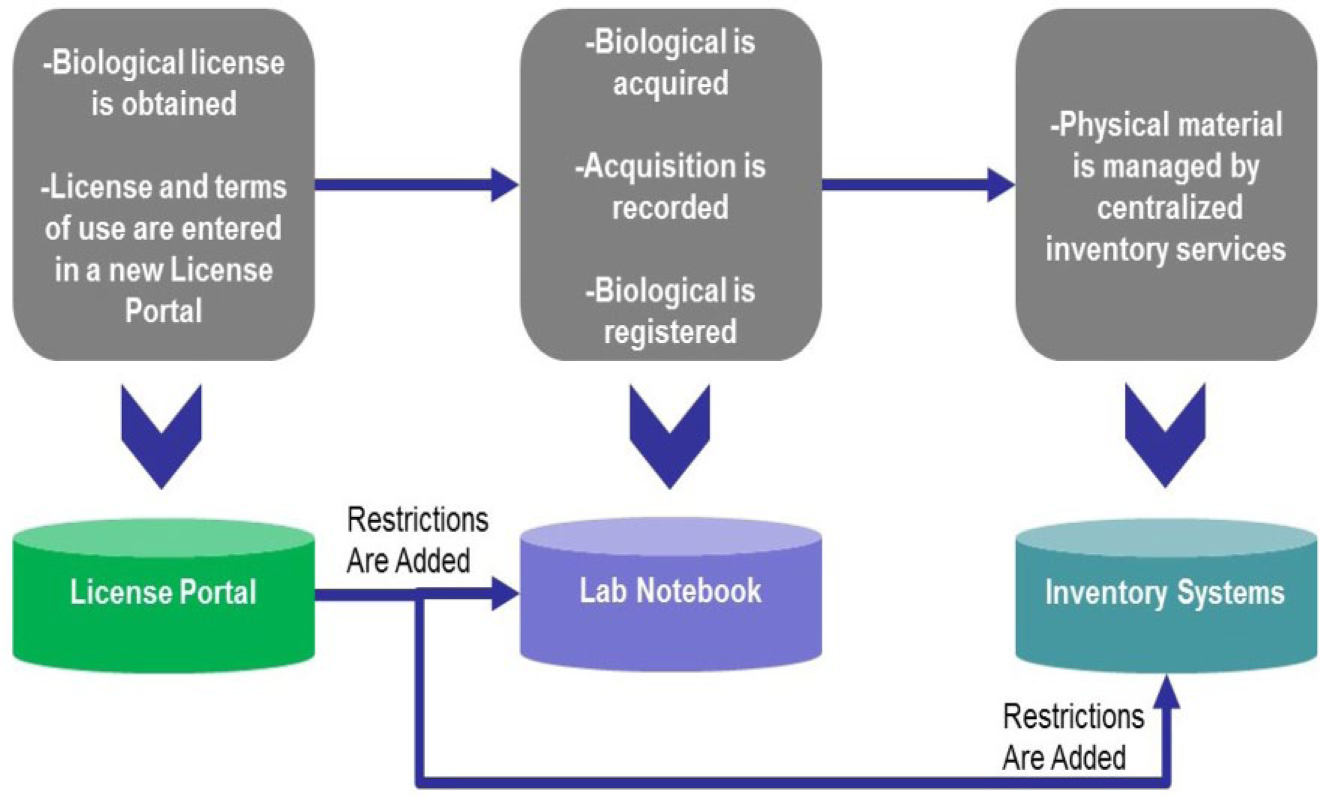
Process for managing biolicenses.
Biological Registration
Biological sample registration ensures the visibility of biological materials. The system assigns a unique identifier to a sample so that it is referred to in the same way across all parts of the organization and enables traceability of the biological entity from creation or acquisition to development and marketing of the drug (in the case of a biological therapeutic). The assignment of the unique ID is normally a prerequisite to submitting the sample into an inventory and allows scientists to search for registered GSK biological materials, minimizing the overlap of purchased and licensed biological materials. At the registration stage, GSK not only collects data on biolicensing but also requires origin and date of acquisition information for other important compliance checks (see the Nagoya section of this review).
Biolicensing Portal
The second measure implemented at GSK is the creation of a data restriction platform (biolicensing portal) that enables the central storage of licenses and their terms. This platform, maintained and managed by subject matter experts, translates complex contractual language into key information accessible to the scientists. Before starting a research project, scientists can check the existence of a license or agreement governing the use of a biological or a technology, any research application restrictions, any geographical restrictions, and any other specific contractual requirements. The biolicensing portal is also essential in monitoring the life cycle of a license by giving clear visibility of the royalty’s payment terms, license expiry, or termination terms.
The biolicensing portal is a key element of a future-state vision to establish automated and integrated internal control processes that enable the management of the physical sample and data according to their associated contractual restrictions.
Nagoya
The Convention on Biological Diversity (CBD) was introduced in 1992 and addressed the sustainable use and conservation of natural resources. 30 The NP 31 is a supplementary protocol to the CBD, and its goal is to provide a transparent and legal framework for the effective implementation of one of the pillars of the CBD, namely, access to and benefit sharing from the use of genetic resources (GRs) of nonhuman origin and associated traditional knowledge (TK) ( Fig. 4 ). The NP entered into force on October 12, 2014, with more than 100 ratifications so far, and more countries are expected to continue this trend. Human GRs are not covered by the scope of the CBD and the protocol and are therefore out of scope of associated regulations.
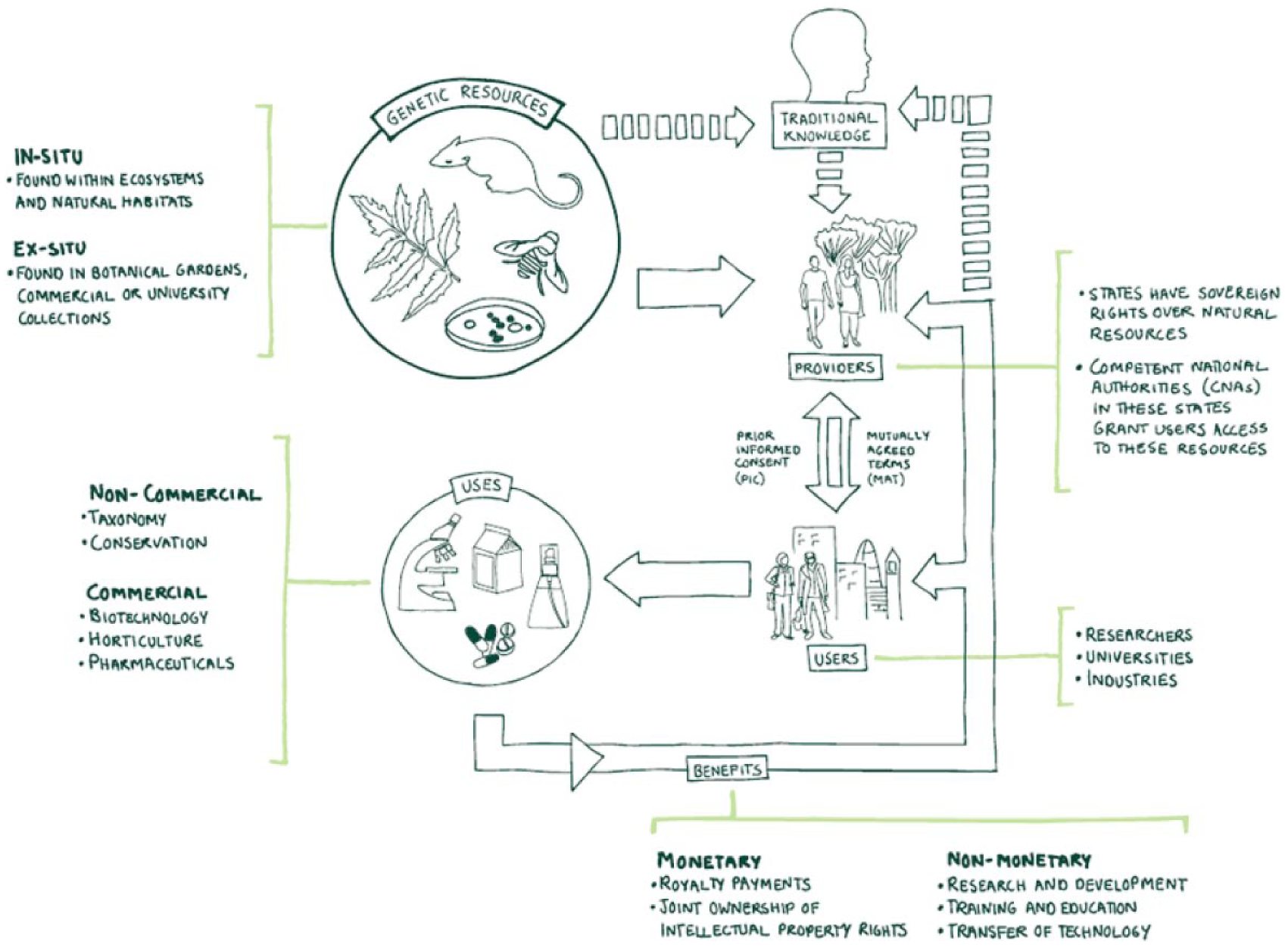
Key themes in ABS: the CBD and Nagoya.
Access and benefit sharing (ABS) legislation is developing rapidly, and as of April 2018, 71% of countries that have signed up to the protocol have published measures (surrounding access of GRs) and legislation (to enforce these measures). 32 These measures are quite diverse, with some countries requiring prior informed consent (PIC) before use of the material in research. Penalties for noncompliance can include large fines, seizure of goods, delays in drug filing or launch, and even custodial sentences.
The European Parliament implemented Regulation (EU) No. 511/2014 on April 16, 2014, on the compliance measures relating to the NP, covering access to GRs and the fair and equitable sharing of benefits arising from their utilization. 33 Although the United States has not signed up to the NP, organizations still have a responsibility to comply with all national laws, and as such, U.S. R&D is subject to foreign ABS requirements.
The fundamental principles of CBD and Nagoya are to protect natural resources and ensure appropriate ABS. These principles should be applied in a way that supports, not undermines, biomedical research. Few pharma or commercial suppliers have published policies on Nagoya, and most organizations are in the earlier phases of developing their procedures and risk mitigation strategies to address these issues.34–36 For those that have shared information, most state that their organization will exercise due diligence to ensure that GRs are accessed and used according to applicable ABS legislation of provider countries.
The NP has implications for the public health response to infectious diseases, including influenza. In February 2017, Canada, France, Germany, Italy, Japan, Mexico, the United Kingdom, the United States, and the European Commission (collectively, the GHSI) 37 voluntarily agreed to facilitate the rapid sharing of noninfluenza biological materials among themselves during a potential or actual public health emergency. It is imperative that in our goal to address global health issues, for example, pandemics, suitable agreements and considerations are established for the rapid access and use of key biological materials to discover and develop effective treatments. There is evidence that certain countries are withholding access to key pathogenic strains, and despite international agreements, U.S. research agencies have not received H7N9 avian flu specimens from their Chinese counterparts. 38 The H7N9 virus is a strong contender as the next global pandemic, and consequently, this is of grave concern to the international community.
All research organizations utilize large numbers of biological materials, and within GSK our scientists are accessing more than 10,000 of these materials each year. Within pharma R&D at GSK, we are implementing many approaches to managing the risk and addressing compliance with the use of such materials. This includes mandating the registration of all biologicals used within R&D, which enables our organization to track and trace GRs through the discovery and development pipeline. This is analogous to our small-molecule registration strategy, which has been in place for many years. Other measures include analysis at key research stage gates to ensure that material utilized in the discovery and development of medicines is documented and any ABS obligation is identified.
Substance Restriction Service: A GSK Application That Ensures Our Inventories Are Fully Compliant
As can be seen from the above discussions, restrictions on a sample can come from varying sources, and it is imperative that these are tracked against both small molecules and biological assets. GSK has designed an application, the Substance Restriction Service, that acts as a repository for all licensing, regulation, and postcandidate assets, as well as other in-house-defined tagging required for samples, and is as diverse as alliance ownership, safety information, and solubility. Where possible, legislation information is pulled directly from our commercial software suppliers and other information from master data sources within GSK or manually entered. The lists from the application are run against our inventories on a regular basis and results are passed through to various downstream systems as required. When Schedule 1 legislation is updated, the Substance Restriction Service will pick up the notification from our commercial package and send the information along to the appropriate downstream applications, for example, inventory and shipping software. Newly registered samples are also checked against the Substance Restriction Service lists to ensure that appropriate tagging is in place. The Substance Restriction Service provides a robust tool to enable navigation of the increasingly complex compliance world.
Shipping
The movement of samples, both chemical and biological, is a critical function within a research organization. Sample management organizations, regardless of their size, often manage large numbers of samples spread across different sites and even different countries. Members within the research community often request sample management’s help to move their materials, both internally and externally, to navigate the ever-growing outsourcing, globalization, and external partnership world. 39 To do this effectively, sample management groups need to utilize the services of external shipping companies and liaise effectively with internal groups, including expert customs, compliance, legal, and employee health departments.
Throughout the shipping process, there are several stages during the transportation, specifically with international shipments: packaging and labeling, ground transportation, customs departure, air transportation, receiving country’s customs, and ground transportation for delivery to final customer ( Fig. 5 ). 40 All paperwork must be correct and in place prior to shipping to allow a smooth transition from one step to the next. To oversee all the movement of these samples and to aid in the regulations, GSK has created several tools and partnered with groups (internal and external) to ensure that all shipments are compliant.
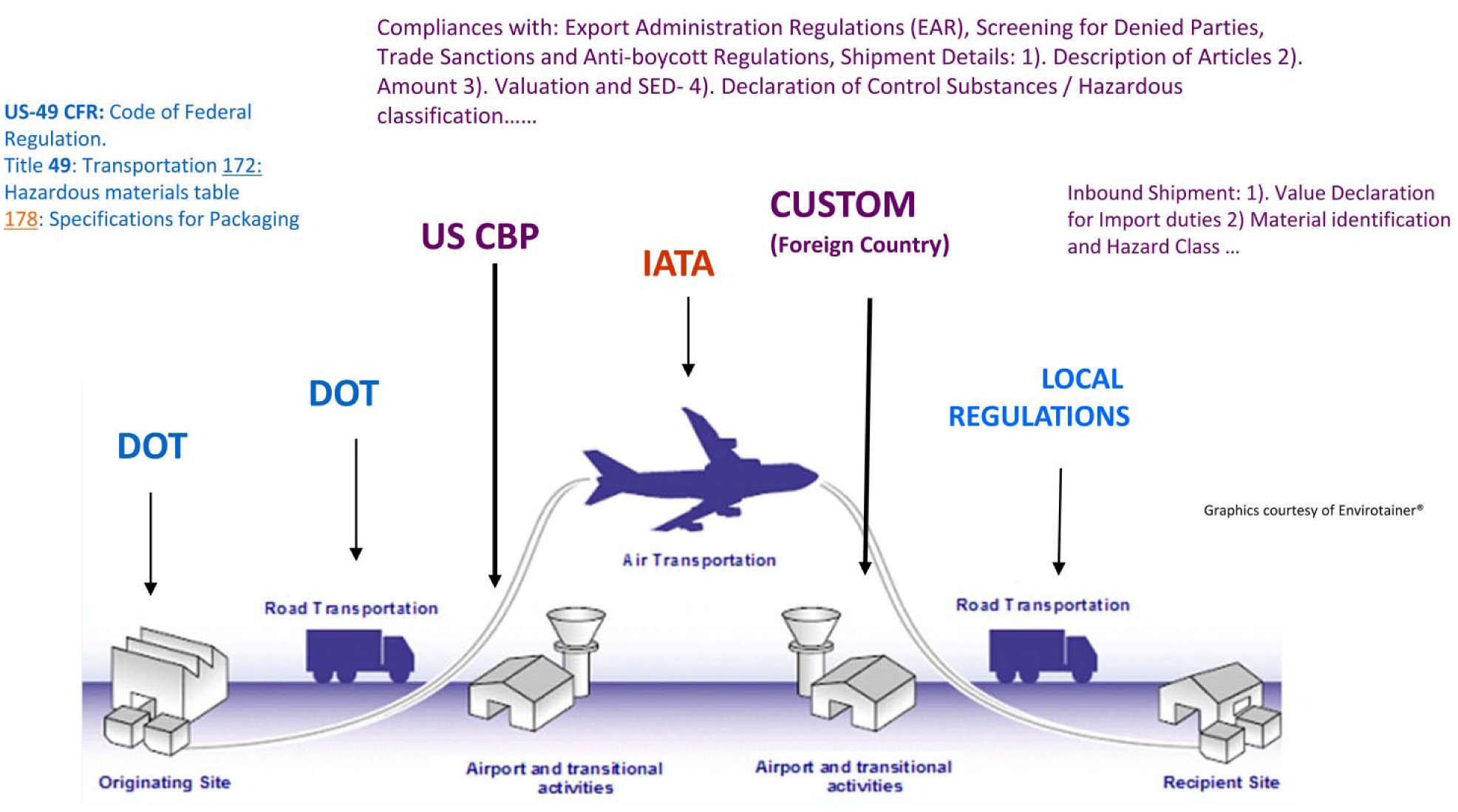
Example of logistical and compliance challenges in supply chains. CBP = Custom and Border Protect Agency; DOT = Department of Transportation; IATA = International Air Transport Association; SED = shipper’s export declaration. 40
GSK has created a shipping application, internally known as GSK Shipper, that aids in the safe transport of materials according to global regulations. This system can be accessed both internally within GSK and externally with secure access. It provides real-time updates, supports the traceability of shipments from initial request to final receipt, assists in monitoring of carrier performance to ensure compliance, and has reporting capabilities. No additional software is required, which drastically reduces the impact on local IT support. The training to use GSK Shipper is minimal with guided screens and built-in help. The person entering the shipment describes what they are sending by filling out a series of mandatory fields, such as type of material, volume or amount, and container the sample is in ( Fig. 6a,b ). Having these mandatory data fields ensures standard and consistent data for record keeping and compliance. This information is transferred automatically to the third-party specialist courier, who uses additional GSK tools, such as a valuation tool, to calculate the valuation of the shipment for import duty costs. By using GSK Shipper, these third parties do not need to rely on the manual transfer of information, which is time-consuming and error-prone.
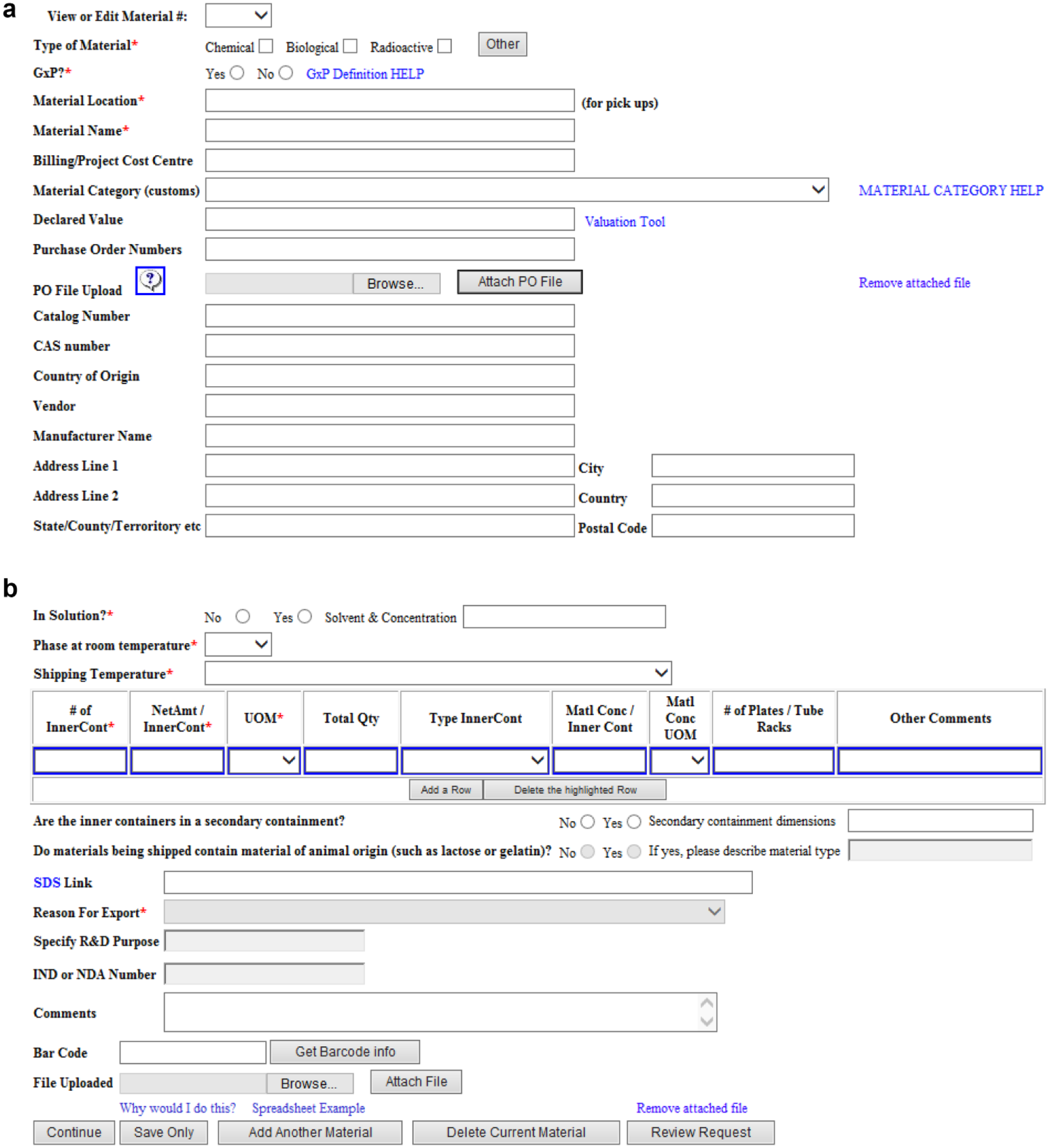
(
Third-party vendors also play an import role ensuring that the proper licenses are in place when shipping biological samples internationally, for example, ensuring the appropriate labeling of shipments if any genetically modified organisms (GMOs) are being shipped, or documentation if any fetal bovine serum is being sent, as there are regulations governing the shipping of such materials. Within the shipping tool, the requestor lists the material and specific information regarding the exact material being sent. This places the responsibility on the individual expert making the request to ensure that the information is correct and not erroneously transcribed. A check is also required for licensing agreements and permission to share the material. If scientists cannot answer the questions, they are then referred to a biolicensing group and the shipment is held until the information is made available. These attributes and checks need to be made by organizations shipping small molecules and biologicals regardless of the specific tools used.
It is also necessary to check for prohibited materials such that country legislation is followed and to prevent shipments from being delayed. Another key parameter is the Harmonized Commodity Description and Code System. Sample management groups need to work with tariff classification specialists who generate this six-digit number known internationally to classify commodities being shipped. 41 In certain circumstances, classifications can be made under the prototype ruling. 42 The prototype HTS number 9817.85.01 is used exclusively for development, testing, product evaluation, or quality control purposes. Under this, organic compounds for pharmaceutical research can be classified as prototypes under Heading 9817.85.01, HTSUS.43,44 The shipment of research materials under this ruling needs to adhere to specific guidelines, and detailed records of all shipments need to be made, together with a record of use for research purposes only.
Due to the international nature of research, it is critical that the transport of research samples is fast, efficient, and compliant. With the increasing cost of drug discovery, organizations can look to reduce costs by combining shipments and reducing their frequency. This, however, can produce potential delays due to an increase in customs holds and review as the value per shipment is increased. This could also impact research cycle times, as the shipping frequency would not match a program’s assay timings. These factors should be taken into consideration to ensure that the sample management supply chain is balanced with the program team’s requirements. Ensuring that shipments have the right classification and description and follow the appropriate compliance checks is the best approach to fast and efficient cycle times and secure deliveries. This in turn requires close collaboration between a sample management expert and many key specialists across an organization.
In the absence of a shipment tool such as GSK Shipper, the process is very manual, the interfaces with third parties are inefficient and time-consuming, and there is no visible chain of custody for audit purposes. GSK Shipper provides interfaces with third-party vendors and real-time tracking of shipments, and it automates the transfer of shipping details to enable fast and efficient cycle times. This information includes the valuation and classification of materials to ensure efficient and compliant custom clearance.
Summary
Organizations face several legal, regulatory, operational, and strategic risks that come with being in a highly regulated industry. Establishing a framework and tools to ensure compliance and manage these risks is fundamental. A framework should define the essential elements expected of compliance and risk management programs and should include written standards and controls, training, communication, management monitoring, and enterprise oversight.
Sample management organizations face significant challenges in the breadth of types of material they manage, the increased collaborative and externalized nature of research, and the changes in legislation and governance applied to the samples they use. Consequently, the most critical compliance elements for sample management groups to address are to understand what materials they have, what restrictions apply and how, and where those materials are used. It is therefore fundamental to register and track the use of samples and be able to link the samples to important metadata regarding restrictions. Information can be tracked manually, but IT applications provide tools to link information across multiple sources and enable and simplify compliance checks at acquisition, experimentation, or transfer.
Sample management groups have always attracted a wide range of skill sets to support their operations. This review has highlighted the need for compliance skills to be added to that list of capabilities. Ensuring that team members have the appropriate development in the compliance arena and that they build good networks with groups such as risk management, legal, and cross-border shipment are key to ensuring the successful support of current drug discovery business needs. Many of the compliance areas described here are not just sample management issues but are enterprise issues that impact R&D and critically need to be addressed in our organizations. Sample management groups should continue to take the lead in establishing enterprise compliance processes and tools for the benefit of their organizations.
Footnotes
Acknowledgements
The authors wish to thank the many individuals and departments across GSK that support discovery supply in their compliance efforts.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.