
Abstract
Tauopathies are neurological disorders characterized by intracellular tau deposits forming neurofibrillary tangles, neuropil threads, or other disease-specific aggregates composed of the protein tau. Tauopathy disorders include frontotemporal lobar degeneration, corticobasal degeneration, Pick’s disease, and the largest cause of dementia, Alzheimer’s disease. The lack of disease-modifying therapeutic strategies to address tauopathies remains a critical unmet need in dementia care. Thus, novel broad-spectrum tau-targeted therapeutics could have a profound impact in multiple tauopathy disorders, including Alzheimer’s disease. Here we have designed a drug discovery paradigm to identify inhibitors of the pathological tau-enabling protein, MSUT2. We previously showed that activity of the RNA-binding protein MSUT2 drives tauopathy, including tau-mediated neurodegeneration and cognitive dysfunction, in mouse models. Thus, we hypothesized that MSUT2 inhibitors could be therapeutic for tauopathy disorders. Our pipeline for MSUT2 inhibitory compound identification included a primary AlphaScreen, followed by dose–response validation, a secondary fluorescence polarization orthogonal assay, a tertiary specificity screen, and a preliminary toxicity screen. Our work here serves as a proof-of-principle methodology for finding specific inhibitors of the poly(A) RNA-binding protein MSUT2 interaction. Here we identify 4,4′-diisothiocyanostilbene-2,2′-sulfonic acid (DIDS) as a potential tool compound for future work probing the mechanism of MSUT2-induced tau pathology.
Introduction
The brain protein tau, a natively unstructured protein encoded by the MAPT gene, performs an important physiological role in neurons by binding to and modulating neuronal microtubule stability.1–3 This activity helps to support the extensive processes neurons extend to conduct neuronal chemical and electrical signaling through axons.4,5 Under neuronal stress or in disease states, tau is often hyperphosphorylated or altered by other posttranslational modifications (PTMs), resulting in a propensity to self-associate and produce detergent-insoluble protein aggregates, including paired helical filaments and neurofibrillary tangles (NFTs).6,7 Neurons exhibit complex patterns of tau expression with multiple splice isoforms and a myriad of PTMs controlling tau function.8–10 Tau deposits may take many pathological forms depending on the associated disorder. Tauopathies, or disorders with primary insoluble tau deposits as hallmarks, include Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, chronic traumatic encephalopathy, and globular glial tauopathy.11,12 The distinct morphology of tau inclusions, molecular tau species, and the brain regional distribution of tau-containing lesions differentiate tauopathy disorder subtypes. 13 For example, Pick’s disease pathology is primarily composed of spherical, silver-positive aggregates composed of the 3R tau isoform (Pick bodies), 14 while progressive supranuclear palsy consists of NFTs and neuropil threads composed of the 4R tau isoform.15,16 Alzheimer’s disease, the primary cause of dementia worldwide, is a complex syndrome and hallmarks include NFTs, neuropil threads, and tau-containing neuritic plaques.12,17 There are no disease-modifying therapeutics for ameliorating pathological tau; therefore, new mechanistic targets and therapeutic strategies for these disorders are desperately needed.1,18–20 Novel therapeutics against targets controlling tau deposition could potentially impact several disorders as tauopathies share common pathologies.
It has been difficult to target tau directly; however, recent work has provided a role for the RNA-binding protein (RBP) MSUT2 in specifically exacerbating the development of toxic tau aggregates while having no obvious effect on total tau levels.21,22 In a mouse model of tauopathy (PS19), MSUT2 overexpression induces pathological tau deposition, widespread hippocampal neuron loss, and deficits in cognition. 22 Knockout of MSUT2 is innocuous on its own, but in the tauopathy background leads to preservation of neurons and cognition by reducing tangle formation and neurofibrillary-mediated neuron loss. MSUT2 binds poly(A) RNA and plays a role in mRNA transcript maturation.23–25 Further, we have previously shown that the specific removal of the MSUT2 RNA-binding domain (CCCH domains) in mice is sufficient to alleviate tau pathology, including tangle deposition in PS19 mouse models. 22 Given this, we hypothesize that targeting the poly(A)–MSUT2 interaction with small molecules will reduce toxic tau burden and may be a viable therapeutic approach for tauopathies.
MSUT2 belongs to a class of proteins known as RNA-binding proteins (RBPs). Considerable evidence has implicated many different RBPs in diverse neurodegenerative diseases. The early-onset neurodegenerative disorder spinal muscular atrophy (SMA) is caused by loss-of-function mutations in the survival of motor neuron 1 (SMN1) gene. 26 SMN1 encodes the SMN protein crucial for transcriptional regulation and the disease-associated loss-of-function mutations cause widespread disruption in splicing homeostasis and are responsible for SMA pathology. 27 Gene therapy targeting SMN1, onasemnogene abeparvovec, has been a tremendous success story in treating RBP-mediated neurodegeneration. 28 Other RBPs implicated in neurodegeneration include TDP-43 in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia,29,30 FUS in ALS, 31 PABPN1 in oculopharyngeal muscular dystrophy, 32 and PARK7 (DJ-1) in Parkinson’s disease, among many others.33–35
To date, there are no clinically approved therapeutics for the treatment of tauopathies, and the only approved treatment modalities, including cholinesterase inhibitors, ameliorate symptoms.1,20 However, there is an ongoing initiative to develop tau-modifying therapeutics that target tau propagation, tau aggregation, tau levels, and tau PTMs, among others.18,36 Targeting MSUT2 presents a challenge because no solved protein structure exists and the precise pathological mechanism of MSUT2 molecular action in tauopathies remains unclear. Further, MSUT2 exhibits no enzymatic activity and its interaction with poly(A) likely comprises a relatively large interacting surface area typical of most RBPs.37,38 MSUT2 also interacts with a critical regulator of mRNA poly(A) tails, PABPN1. Thus, therapeutic strategies must specifically target MSUT2 without disrupting PABPN1 function because PABPN1 is essential. 22 Finally, the potentially challenging nature of RBPs as screening targets has resulted in relatively few small-molecule screening initiatives to date.
Despite these challenges, here we describe a discovery pipeline capable of identifying small molecules that specifically disrupt poly(A)–MSUT2 interaction. Our strategy incorporates a primary AlphaScreen, dose–response validation, orthogonal fluorescence polarization (FP) assay validation, a specificity counterscreen against PABPN1, and a cellular toxicity screen. We screened the Spectrum Collection as a proof-of-principle application of our discovery pipeline. We chose the Spectrum Collection to develop our assay for its diversity in that it includes both small molecules and natural products, while also being composed of known biologically active compounds. Our screening effort identified 4,4′-diisothiocyanostilbene-2,2′-sulfonic acid (DIDS) as a potential tool compound, and our screen has demonstrated the ability to find specific and potent compounds targeting the poly(A)–MSUT2 interaction with future larger drug discovery efforts.
Materials and Methods
RNA
5′ biotin-labeled and unlabeled poly(A) RNA were purchased (IDT, Coralville, Iowa; sequences 5′ biotin-AAAAAAAAAAAAAAA-3′ and 5′-AAAAAAAAAAAAAAA-3′). Both biotin-labeled and unlabeled RNA were diluted to 100 µM in RNAse/DNAse free Qiagen water and stored at −80 °C, away from light. RNA was diluted to a working concentration in Alpha Assay buffer just before screening.
Recombinant Protein
MSUT2 ZF and PABPN1 cDNAs were cloned into the pGEX-6P1 vector (Pharmacia, Uppsala, Sweden). MSUT2 ZF- and PABPN1-encoding plasmids were transformed into BL21 (DE3) bacteria. Ten milliliters of Terrific Broth (TB) starter cultures were grown overnight at 37 °C in a shaking incubator. The following morning, 1 L of TB cultures were inoculated and grown at 37 °C with shaking to log phase and induced with a 1 mM final concentration of IPTG for 4 h at 37 °C. Bacterial pellets were resuspended in lysis buffer (phosphate-buffered saline [PBS], 0.3% lysozyme, 1 mM DTT, 1.5% Sarkosyl, 0.5 mM phenylmethylsulfonyl fluoride (PMSF) RNAse A, and DNAse I). Affinity-based gravity column purification was performed by binding glutathione S-transferase (GST)-tagged MSUT2 or PABPN1 to sepharose-glutathione resin and subsequently eluting with GST elution buffer (20 mM glutathione, 100 mM Tris, pH 9.5). The resulting eluate was buffer exchanged into storage buffer (PBS) and stored at −80 °C. The protein purity and yield were analyzed via Bradford assay and Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Chemical Library and Powder Stocks
The Spectrum Collection was purchased from MicroSource (Gaylordsville, CT) and contained a total of 2000 compounds in DMSO. For screening, 50 nL of compound was transferred via a pin tool to a final screening concentration of 33 µM. Initial dose–response curves were generated from cherry-picking library plates. Powder stock-based hits that were retested and passed these screens were purchased from Sigma (St. Louis, MO) (2,3-dichloro-5,8-dihydroxynapthoquinone [Sigma 343420], 4,4′-diisothiocyanostilbene-2,2′-sulfonic acid sodium salt [Sigma D3514], aurin tricarboxylic acid [Sigma A36883], and ebselen [Sigma E3520]) and used in generating figures (see
AlphaScreen Assay
Samples were set up in 384-well PerkinElmer (Waltham, MA) white opaque-bottom plates (PE06). All reactants were diluted in Alpha Assay buffer (1× AlphaLISA universal buffer [part AL001] + 100 mM NaCl). A total reaction volume of 15 µL was plated in the 384-well format using a CyBio (Jena, Germany) Well Vario (6 µL of donor beads [10 µg/mL], 3 µL of biotinylated RNA [250 nM], and 3 µL of GST-MSUT2 protein [250 nM] per well). Fifty nanoliters of compounds were transferred to wells via the CyBio Vario equipped with a pin tool. This mixture was incubated at room temperature for 30 min away from light. Next, 3 µL of acceptor beads (1.25 µg/mL) were added. Plates were incubated at room temperature away from light for 60 min and subsequently read on a PerkinElmer EnVision multimode microplate reader equipped with stackers using a standard 384-well Alpha Assay software protocol.
Fluorescence Polarization Assay
A follow-up FP assay was performed in a half-area black plate (Corning, Corning, New York, 3686). Fifty microliters of 125 nM MSUT2 and 10 nM FAM-RNA (ordered from IDT) in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4) was transferred using an Integra (Hudson, NH) Viaflo with a 96/50 µL head. Two microliters of compound was transferred for a final concentration of 10 µM. Plates were incubated for 20 min at room temperature away from light and read using a Cytation 5 (BioTek, Winooski, VT) with a preconfigured green polarization filter cube (Biotek, 8040561) at excitation 485/20 and emission 528/20, a dichroic mirror at 510 nm, and a read height of 10 mm. FP was calculated by first subtracting the background from a buffer-only control well and then using the equation
Cell Culture and Cytotoxicity Screen
HEK-293 was cultured with cell growth medium (Dulbecco’s modified Eagle’s medium, 10% defined fetal bovine serum, penicillin [1000 IU/mL], and streptomycin [1000 µg/mL]) as described. 22 Cell viability was assessed using Promega CellTiter-Glo (Promega, Madison, WI, G7570) per the manufacturer’s protocol. Briefly, HEK-293 cells were grown to 70% confluence in a 96-well plate, treated with the indicated concentrations of compounds, and incubated for 72 h at 37 °C. Plates were read for luminescence on a PerkinElmer EnSpire Alpha.
Statistical Analyses and Figures
Four-parameter nonlinear regression was used to calculate the IC50 values generated via GraphPad Prism 8 (GraphPad, La Jolla, CA).
Results
AlphaScreen Design and Underlying Principle
AlphaScreen technology relies on donor beads, coated with a hydrogel excitable by 680 nm of light, brought within close proximity to an acceptor bead leading to light emission and detection. In our assay, streptavidin-coated donor beads tightly bind to biotinylated-poly(A) RNA, while the glutathione-coated acceptor bead binds to GST-tagged MSUT2 protein. Because of the known high affinity of MSUT2 for poly(A) (Kd = 60 ± 15 nM),
22
beads are brought within the needed radius required for the donor bead to excite the acceptor bead via singlet oxygen transference. When this interaction is blocked by an inhibitor, the proximity threshold for donor and acceptor beads is not met, singlet oxygen transference cannot occur, and light is not emitted by the acceptor bead (
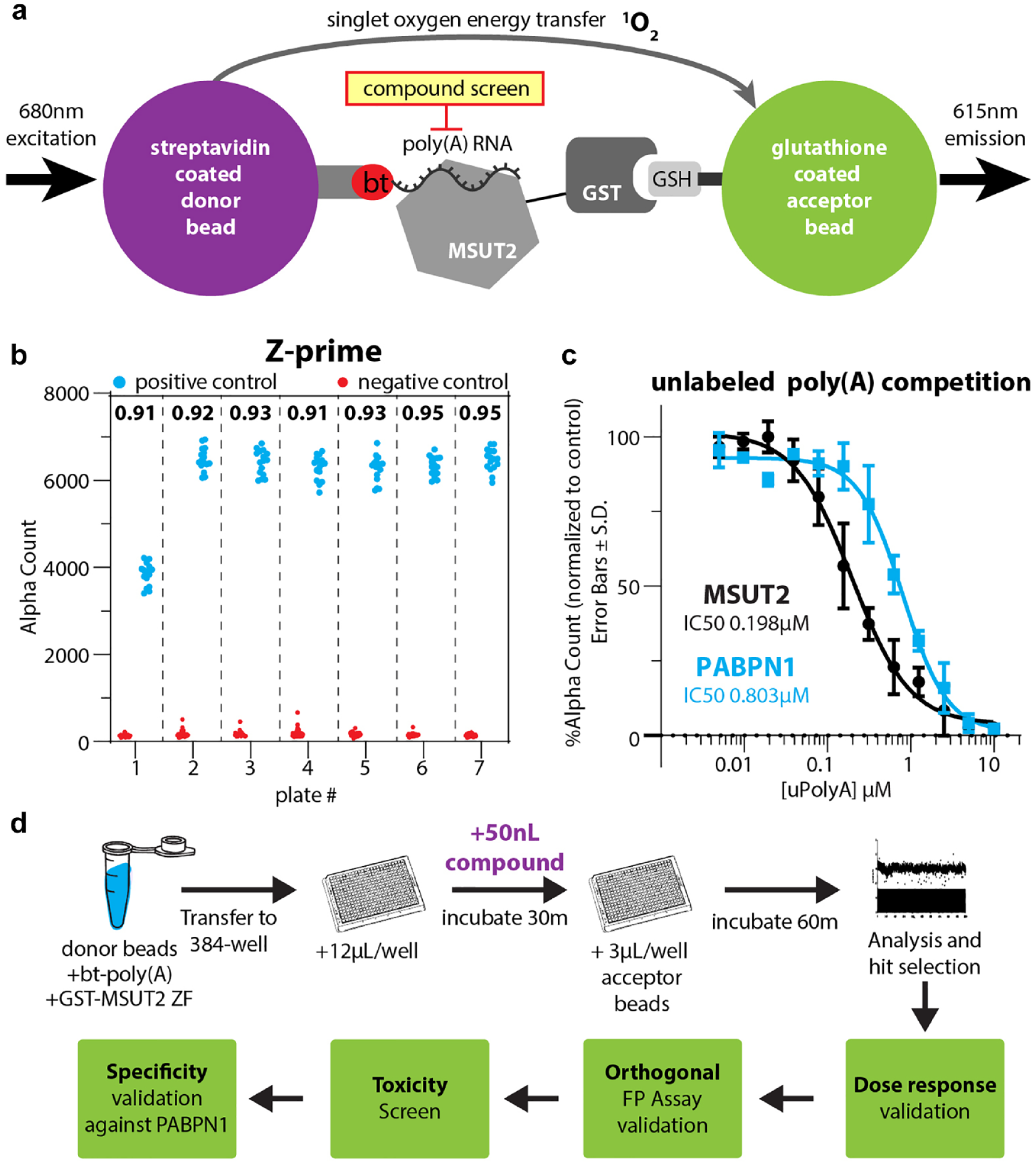
(
Screening of the Spectrum Collection
Screening of the 2000-compound Spectrum library resulted in the identification of 20 initial hits, for a hit rate of 1%. Our hit window consisted of those compounds 7 standard deviations from the mean (
Summary of IC50 Values for the Three Identified Compounds.
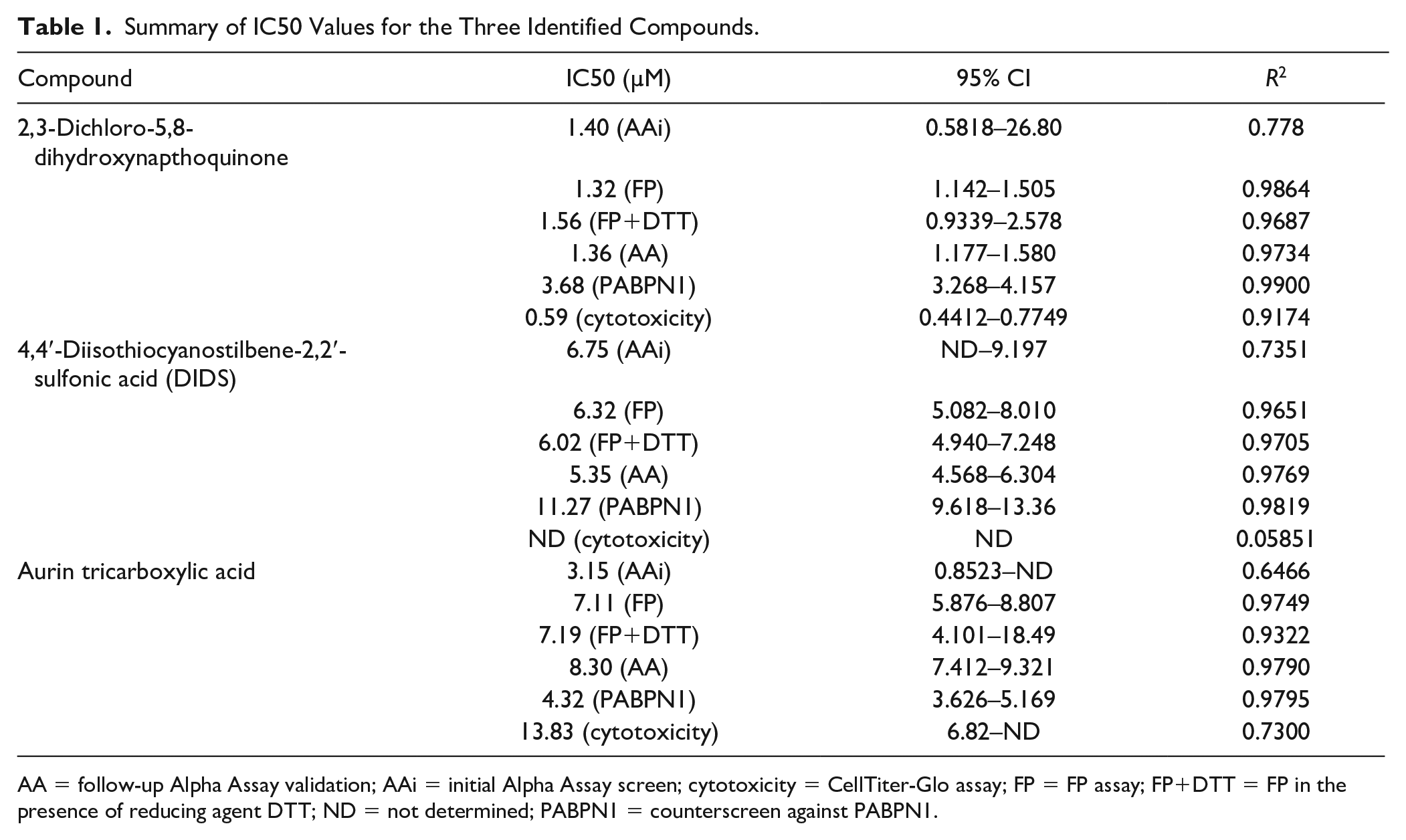
AA = follow-up Alpha Assay validation; AAi = initial Alpha Assay screen; cytotoxicity = CellTiter-Glo assay; FP = FP assay; FP+DTT = FP in the presence of reducing agent DTT; ND = not determined; PABPN1 = counterscreen against PABPN1.
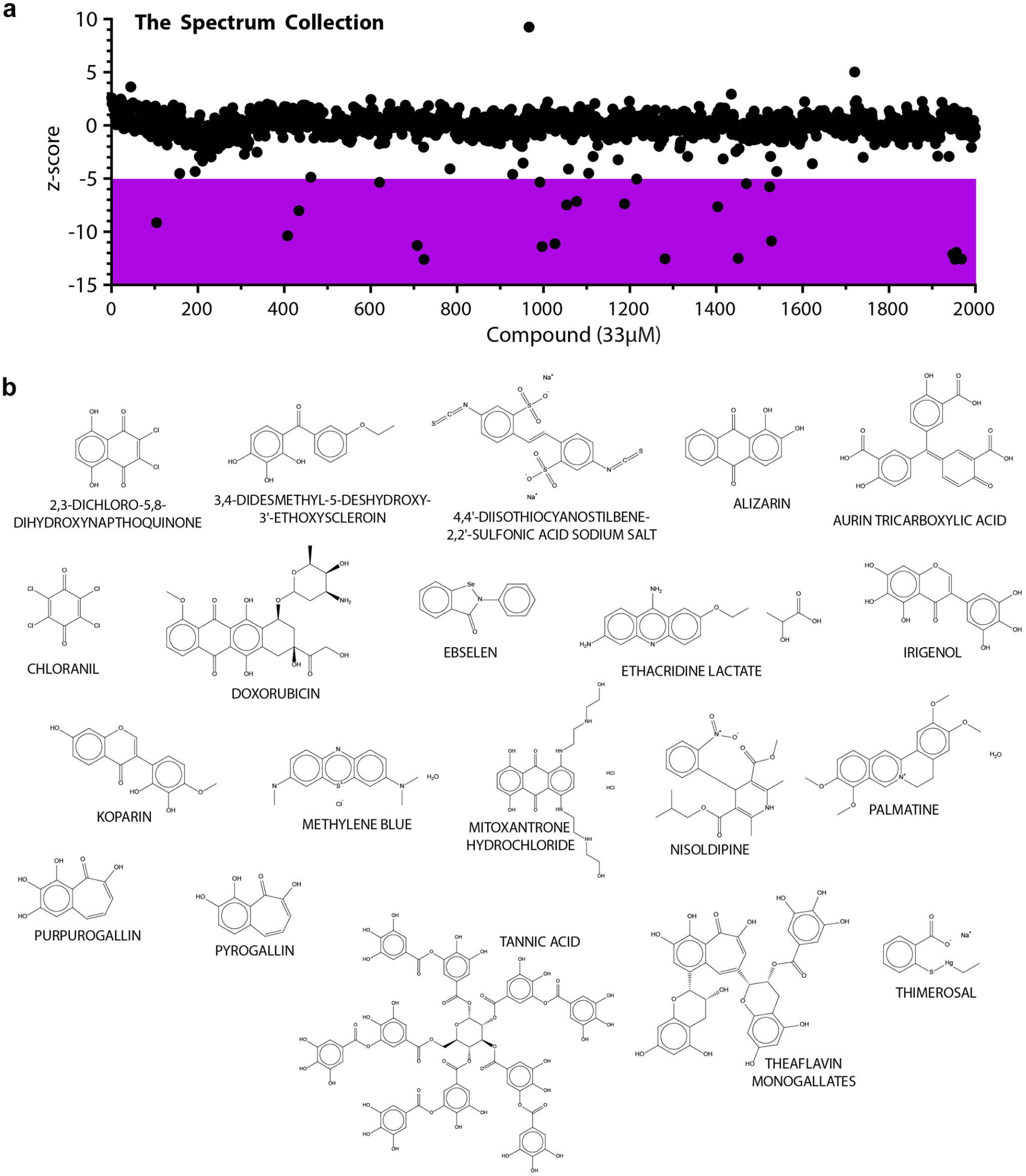
Spectrum Collection AlphaScreen. (
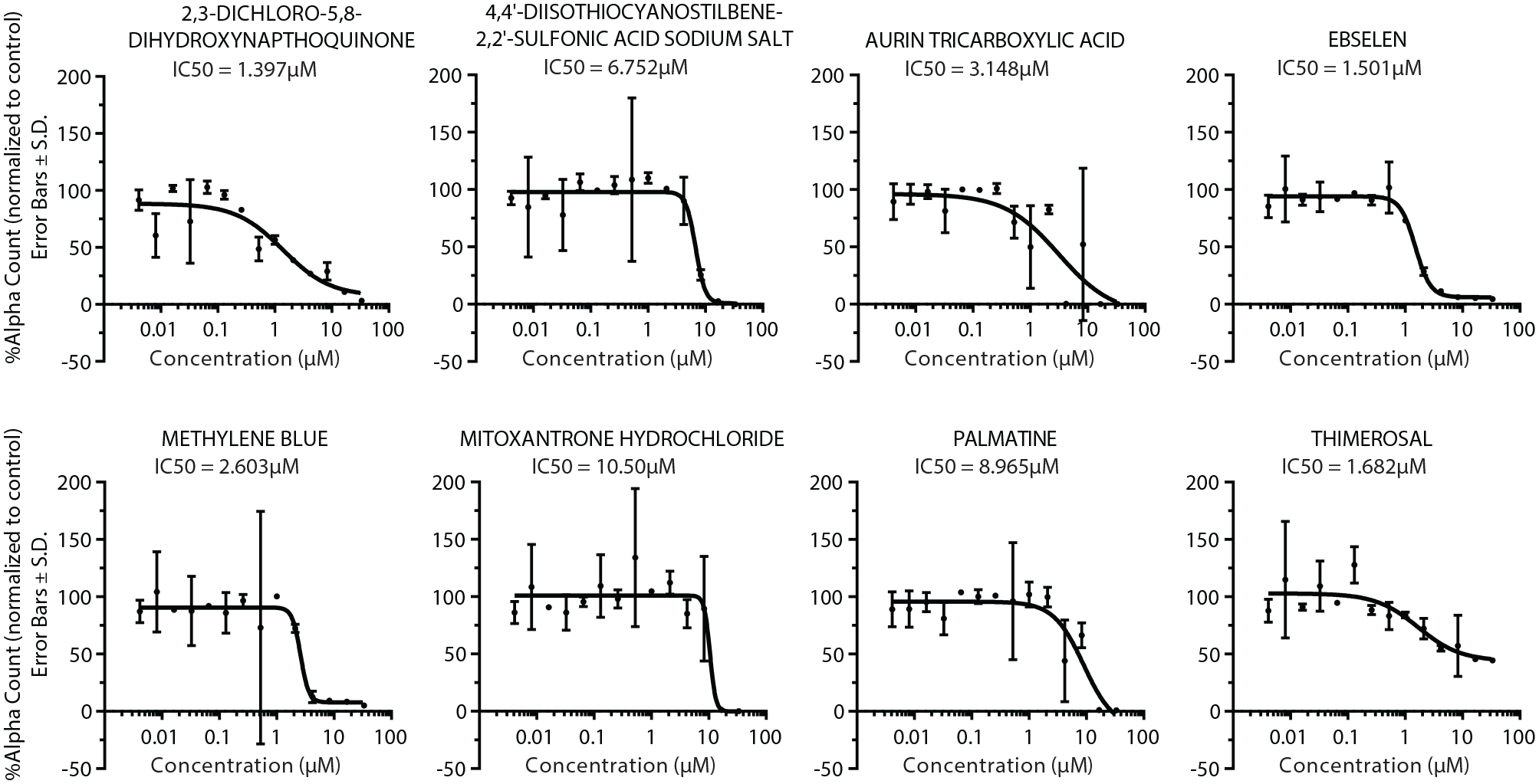
Alpha Assay dose–response validation for hits. The Y axis is the percentage of the alpha count normalized to the DMSO control. Errors bars represent the standard deviation, and N = 3 for each concentration. IC50 is shown for each compound, calculated by fitting to a four-parameter nonlinear regression curve in GraphPad Prism 8.
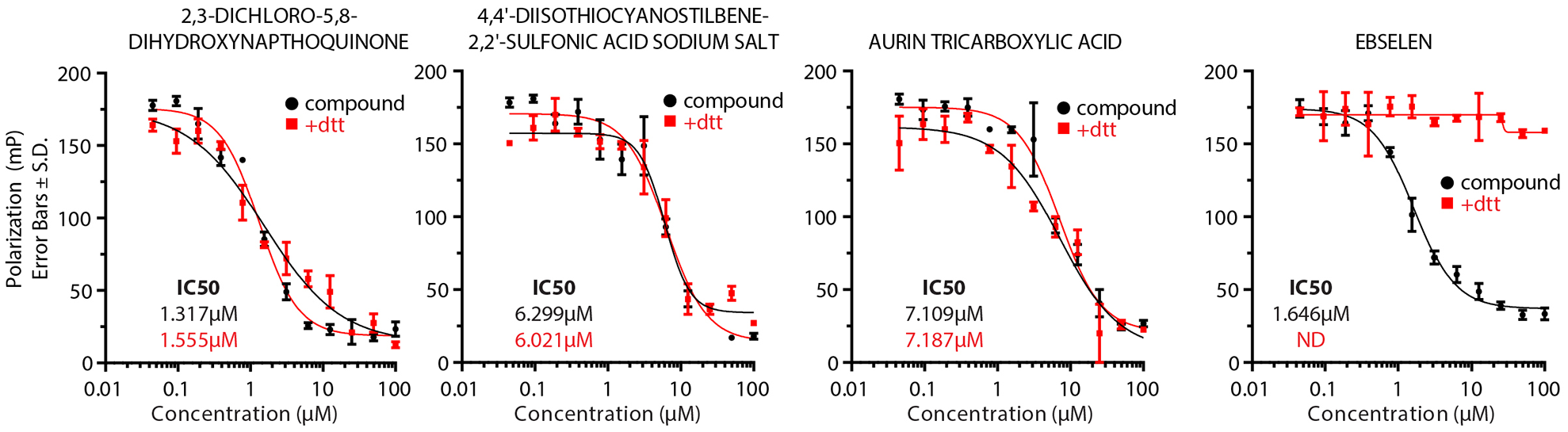
FP orthogonal screen results. MSUT2-bound FAM-poly(A) RNA emits highly polarized light, while inhibition results in free FAM-poly(A) and emits nonpolarized light. The Y axis is the polarization units (mP) and the X axis is the compound concentration in micromoles. FAM-labeled RNA IC50 is indicated for all compounds with DTT (red) or without DTT (black). Error bars are the standard deviation, and N = 3 for compounds without DTT and N = 2 for compounds with DTT.
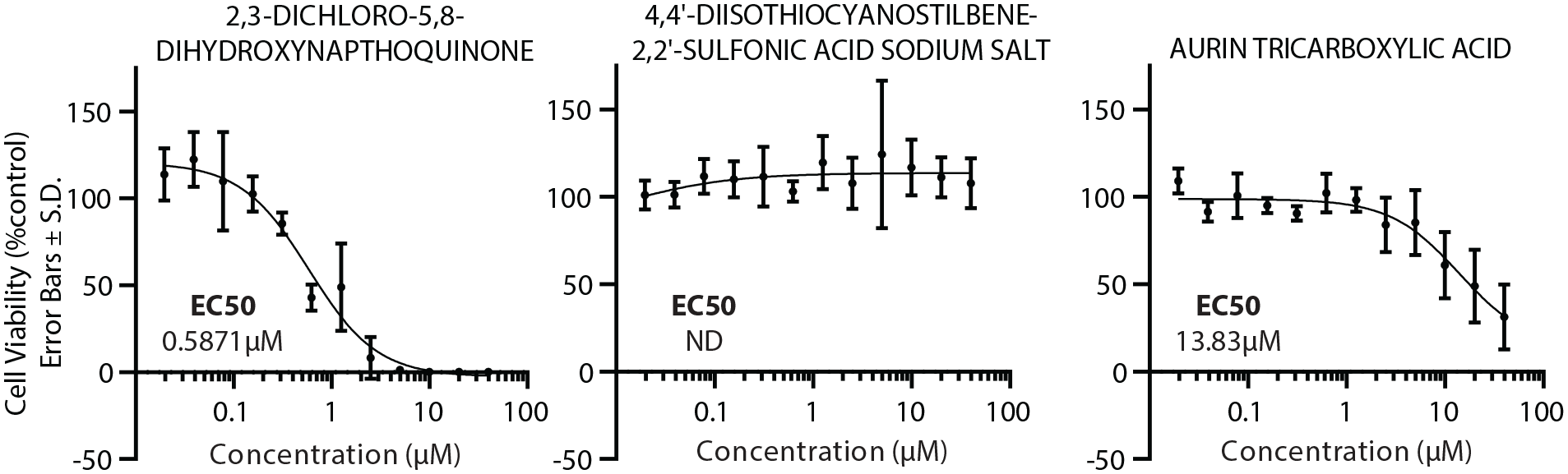
Cellular toxicity assay. Cell viability was measured by CellTiter-Glo (Promega) for three different compound treatments at indicated concentrations and normalized to the control DMSO treatment. Data points consist of two biological replicates, each at N = 3. Error bars represent the standard deviation. EC50 was calculated by fitting to a four-parameter nonlinear regression.
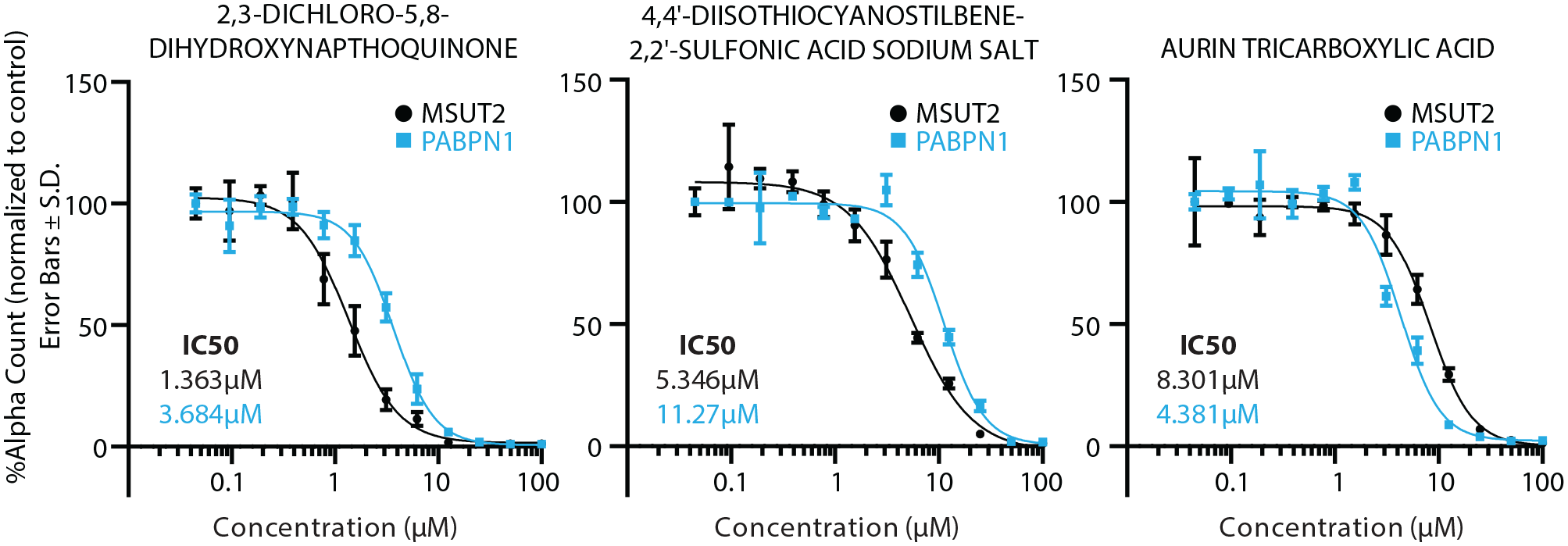
Selectivity screen. Compounds were screened by AlphaScreen for activity against MSUT2 (black) and PABPN1 (blue). Error bars are the standard deviation, where N = 3 for each concentration. IC50 was calculated by fitting to a four-parameter nonlinear regression. The IC50 values against MSUT2 (black) and PABPN1 (blue) are indicated.
Discussion
A tremendous unmet need exists for tau-modifying therapeutics to treat tauopathy disorders. Directly engaging tau shows promise as a strategy for treatment, and there are a number of biologicals, including tau antibodies and antitau vaccines, as well as small-molecule aggregation inhibitors and PTM modifiers, being tested for efficacy in patients with tauopathy disorders (primarily Alzheimer’s disease and progressive supranuclear palsy). 18 Most small-molecule approaches directly targeting tau aggregation or phosphorylation have been abandoned for lack of efficacy or because of off-target complications, and the majority of more advanced investigational therapeutics currently utilize immunotherapy-based modalities. 19 Some tau-targeting immunotherapies, including BMS-986168 (gosuranemab), failed to show efficacy in PSP but are being investigated for AD-mediated mild cognitive impairment (ClinicalTrials.gov ID NCT03068468), and Abbvie’s AADvac1 and C2N-8E12 are also in early-stage clinical trials. 18 Determining which tau species to target has been a challenge because pathological tau encompasses a variety of misfolded or aberrantly modified monomeric tau species, toxic gain-of-function oligomers, paired helical filaments, and NFT deposits.1,42–45
MSUT2 has emerged as an alternative for indirectly targeting pathological tau. It has been shown that knocking out MSUT2 in mice (PS19) overexpressing aggressively aggregating P301S-mutated human tau prevents toxic oligomeric tau and NFT deposits while preserving neuronal health in the hippocampus and memory as evaluated by the Barnes maze paradigm. 22 These mice develop normally and do not have obvious defects as a result of the loss of MSUT2. While it is known that MSUT2 has a role in poly(A) tail length, its precise pathological mechanism of action remains unclear. Mislocalization of MSUT2 from the nucleus to the cytoplasm may provide a toxic gain-of-function activity, allowing it to induce pathological tau formation, but this has not been shown directly. We have also hypothesized that MSUT2 may cause poly(A) RNA, a polyanion, to seed the pathological aggregation of tau. Regardless of the exact mechanism by which MSUT2 leads to tau accumulation, knocking down MSUT2 specifically affects toxic tau species, including oligomers and NFTs.21,22,46
Traditionally, many RBPs and specifically the RNA-RBP interaction have been considered less than ideal for small-molecule screening campaigns as they lack a targetable enzymatic pocket. However, here we have shown with two screening paradigms the ability to identify compounds that are specific and potent in blocking the poly(A)–MSUT2 interaction. With the application of AlphaScreen and FP technology, high-throughput screening initiatives for RNA–protein inhibitors have been successful for various RBP targets.47–49 The compound identified here, DIDS, was shown to inhibit poly(A)–MSUT2 binding at ~5 µM and had a twofold reduction in potency against PABPN1 (~11 µM), the counterscreen target. We anticipate that higher specificity will be required for translationally effective compounds but believe the identification of DIDS provides the proof of concept required for screening larger drug libraries.
DIDS has primarily been investigated for its use in binding and blocking chloride ion channels in mammalian cell models. Interestingly, it has been found that DIDS is able to inhibit another polynucleotide binding protein, RAD51, a DNA recombinase essential for double-strand break repair in eukaryotes, and has been investigated as an anticancer agent. 50 Although both the potency and specificity are not sufficient to further pursue DIDS for therapeutic potential, it may prove useful as a tool compound in determining molecular mechanisms of MSUT2-induced tau toxicity.
MSUT2 is a promising target in combatting tau-mediated neurodegeneration. Advancing our understanding of fundamental MSUT2-mediated mechanisms of neurodegeneration remains a critical component of therapeutic development. The identification of these initial inhibitors will allow us to probe MSUT2 function and facilitate the development of more translationally suitable MSUT2 inhibitors. Likewise, solving the structure and determining the pathological mechanism of MSUT2 will present new opportunities in targeting MSUT2-induced tau pathology. A solved structure will allow for virtual docking of compounds and more precise medicinal chemistry initiatives for optimizing small molecules, while determining its role in biological and pathological pathways will allow for reducing off-target complications and may provide new therapeutic targets.
Footnotes
Acknowledgements
We thank Timothy Martins and Elaine Loomis for sharing their technical expertise. We thank Timothy Martins, James Annis, and the Quellos high-throughput screening center for sharing advice on assay development, instrumentation, and previous experience.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.D.B., T.J.S., and B.C.K. have a patent application pending (assigned U.S. Patent Application No. 16/383,178), “Compositions and Methods for Suppressing MSUT2.”
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the National Institutes of Health (R56AG057642 and RF1AG055474 to B.C.K.), the Department of Veterans Affairs (Merit Review Grant no. I01BX002619 and VA PSHCS ACOS/R seed funding to B.C.K.), and a postdoctoral fellowship from the Washington Research Foundation (to J.D.B.).
Data Availability
Data is available upon request to the corresponding author (email: