
Abstract
Hematopoietic progenitor kinase 1 (HPK1), also referred to as mitogen-activated protein kinase kinase kinase kinase 1 (MAP4K1), is a serine/threonine kinase that negatively regulates T-cell signaling by phosphorylating Ser376 of Src homology 2 (SH2) domain-containing leukocyte protein of 76 kDa (SLP-76), a critical mediator of T-cell receptor activation. HPK1 loss of function mouse models demonstrated enhanced immune cell activation and beneficial antitumor activity. To enable discovery and functional characterization of high-affinity small-molecule HPK1 inhibitors, we have established high-throughput biochemical, cell-based, and novel pharmacodynamic (PD) assays. Kinase activity-based time-resolved fluorescence energy transfer (TR-FRET) assays were established as the primary biochemical approach to screen for potent inhibitors and assess selectivity against members of MAP4K and other closely related kinases. A proximal target engagement (TE) assay quantifying pSLP-76 levels as a readout and a distal assay measuring IL-2 secretion as a functional response were established using human peripheral blood mononuclear cells (PBMCs) from two healthy donors. Significant correlations between biochemical and cellular assays as well as excellent correlation between the two donors for the cellular assays were observed. pSLP-76 levels were further used as a PD marker in the preclinical murine model. This effort required the development of a novel ultrasensitive single-molecule array (SiMoA) assay to monitor pSLP-76 changes in mouse spleen.
Introduction
HPK1 is a serine/threonine kinase that belongs to the mammalian Ste20-like family. 1 HPK1 was originally cloned as a kinase that can activate JNK and is predominantly expressed in hematopoietic cells in adults despite a widespread expression pattern during embryonic development.2,3 Extensive studies revealed that HPK1 is a negative regulator of T-, B-, and dendritic cell signaling pathways.4–6 The regulatory effect of HPK1 on T-cell signaling is exerted through phosphorylating SLP-76 on Ser376, an adaptor protein that is critical for the signaling pathway of T-cell activation, which leads to subsequent ubiquitination on lysine 30 and SLP-76 degradation. 7 HPK1 can be activated by kinases like LCK and ZAP70 through phosphorylation on tyrosine 379 and can be further activated upon interaction with SLP-76.8–10 In addition to its role regulating SLP-76 phosphorylation, HPK1 can also attenuate T-cell signaling by phosphorylating other interacting adaptor proteins such as GADS at threonine 254. 11 HPK1-deficient primary T cells have enhanced signaling as demonstrated by hyperproliferation and increased Th1 cytokine secretion upon TCR activation. 12 The negative effect of HPK1 on B-cell activation is through phosphorylation of threonine 152 of B-cell linker protein (BLNK) and its subsequent degradation. It is noted that B cells deficient in HPK1 have enhanced capability to produce IgM and IgG.4,5 Dendritic cell activation and maturation is also negatively impacted by HPK1 activation, and bone marrow-derived dendritic cells deficient in HPK1 have enhanced antigen-presenting activity. 6
The complex role of HPK1 in negatively regulating multiple types of immune cells indicates that HPK1 antagonism could promote autoimmunity and may potentially be helpful for antitumor immunotherapy. Ablation of HPK1 in mice renders them more sensitive to the development of experimental autoimmune encephalomyelitis (EAE). 4 Downregulation of HPK1 has been found in T cells of systemic lupus erythematosus patients and PBMCs of psoriatic arthritis patients,13–15 supporting the role of HPK1 in autoimmune pathogenesis.
While immunotherapy with checkpoint inhibitors has changed the landscape of cancer treatment during the past several years, a high fraction of cancer patients still exhibit primary resistance with insufficient initial responses or acquired resistance during the treatment process leading to disease progression. 16 To overcome the immunosuppressive tumor microenvironment, it is critical to break the immune tolerance allowing recognition of tumor antigens. 12 Recent studies demonstrate that antagonizing HPK1 function is likely to enhance immunity toward cancer cells due to improved tumor antigen recognition, increased Th1-type cytokine release due to prolonged T-cell activation, loss of suppressive ability of natural regulatory T cells, and resistance to prostaglandin E2-induced immune suppression.6,12,17–20 Consistent with these observations, HPK1-deficient mice are resistant to Lewis lung carcinoma growth. Furthermore, adoptive transfer of T cells demonstrates that the tumor rejection is T cell mediated. 20 HPK1 kinase-dead knock-in mouse models reported by two independent groups have reached the conclusion that the kinase activity of HPK1 is essential in negatively regulating immune surveillance.21,22 Mice harboring a kinase-dead HPK1 mutant in place of the wild-type (WT) gene have improved T-cell immunity and are resistant to the growth of two tumor models: GL261 glioma and 1956 sarcoma.21,22 Inactivation of HPK1 alone was not sufficient to inhibit the growth of the MC38 tumor model, but it enhanced the effect of anti-PDL1 treatment. 21 These results demonstrated that small-molecule HPK1 inhibitors are potentially beneficial as novel agents for antitumor immunotherapy either as a single agent or in combination with anti-PD1/PD-L1 treatment.
In this study, high-throughput, biochemical, cell-based, and novel PD assays were implemented to support efforts to identify, differentiate, and optimize selective and potent HPK1 inhibitors.
Materials and Methods
Commercial Reagents
Buffers and biochemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. HPK1 kinase (catalytic domain residues 1–346 accession no. NP_009112.1 with an N-terminal DYKDDDDK tag) was purchased from Carna Biosciences, Inc. (Japan). Mitogen-activated protein kinase kinase kinase kinase 3 (MAP4K3) (catalytic domain residues 1–380 accession no. NP_003609.2 with an N-terminal GST tag) was purchased from Thermo Fisher Scientific (Carlsbad, CA). Anti-pSLP-76 (Ser376; D7S1K) Rabbit monoclonal antibody (mAb; AlexaFluor 647 conjugate) was purchased from Cell Signaling Technology (Danvers, MA). Lance Ultra Ulight-p70 S6K (Thr389) and Lance Ultra Europium-anti-phospho-p70 S6K (Thr389) antibody were purchased from PerkinElmer (Waltham, MA). SLP-76 and pSLP-76 Ser376 antibodies, protease/phosphatase inhibitor cocktail, mouse pSLP-76 (Ser376) enzyme-linked immunosorbent assay (ELISA) kit, and PathScan human pSLP-76 (Ser376) sandwich 384-well ELISA kits were purchased from Cell Signaling Technology. AlphaLISA SureFire Ultra p-SLP-76 (Ser376) HV assay kit was purchased from PerkinElmer. pSLP-76 (Ser376) cellular kit was purchased from Cisbio (Bedford, MA). Human IL-2 ELISA plates were purchased from Meso Scale Diagnostics (MSD), LLC (Rockville, MD). Dynabeads Human T-Activator CD3/CD28 and 384 channel reservoirs were purchased from Thermo Fisher Scientific. CellTiter-Glo luminescent cell viability assay reagent was purchased from Promega (Madison, WI). Tissue culture plates (384-well) were purchased from Corning (Corning, NY). RPMI GlutaMAX media, nonessential amino acids, and sodium pyruvate were purchased from Life Technologies (Carlsbad, CA). Hamilton Robotics CO-RE tips were purchased from Hamilton (Reno, NV). Echo LDV source plates were purchased from Labcyte (San Jose, CA). Single-molecule array (SiMoA)-related reagents were purchased from Quanterix Corporation (Billerica, MA).
Cells
Cryopreserved human PBMCs were purchased from Cellular Technology Ltd. (Shaker Heights, OH). Jurkat cells were purchased from ATCC (Manassas, VA). HPK1-deficient Jurkat knockout clones were created by Thermo Fisher Scientific (Waltham, MA). To knock out the human HPK1 in Jurkat cells using the CRISPR/Cas9 technique, the following two sgRNAs were used for targeting exon 3 of the HPK1 gene: sgRNA 1.1, GCCAACATCGTGGCCTACCA TGG (underlined, PAM sequence), and sgRNA 1.3, CCCATGGTAGGCCACGATGTTGG (underlined, PAM sequence). Jurkat cells were transfected with GeneArt Cas9 Nuclease Protein and CRISPR IVT gRNA via a Neon Transfection protocol. Individual clones were isolated from the transfected population via limited dilution cloning, expanded, and screened for HPK1 disruption by PCR amplification of genomic DNA and next-generation sequencing (NGS). HPK1 disruption (indels leading to frameshift and early stop codons) was confirmed in the following clones by another round of NGS: clone B1 from sgRNA 1.1 and clones C11 and D9 from sgRNA 1.3. Mouse spleen T cells were isolated from C57BL/6 mice.
HPK1 Biochemical Assay
All assay solutions were prepared in assay buffer consisting of 50 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.01% Brij-35, 0.05% bovine serum albumin, and 0.5 mM TCEP. Assay solutions were dispensed into a black, 384-well assay plate (Corning 3820) using a BioRPTR (Beckman Coulter, Brea, CA). HPK1 enzyme solution (5 µL, at 1.5 times its final concentration of 75 pM) was added to all wells and the kinase reaction initiated by addition of 2.5 µL of substrate solution (at three times the final concentration of 10 nM SLP-76 protein and 10 µM ATP, or at other concentrations as indicated in the figure legends). Following incubation at room temperature for 1 h, reactions were quenched by addition of 2.5 µL of stop and detection solution (at four times the final concentrations of 10 mM EDTA, 0.75 nM LANCE Eu-W1024 Anti-6xHis Ab, and 0.75 nM pSLP-76 [Ser376; D7S1K] XP Rabbit mAb [AlexaFluor 647 conjugate]). After an additional 1 h of incubation at room temperature, the TR-FRET signal was measured on an Envision (PerkinElmer) plate reader set for 320 nm excitation and dual-emission detection at 615 nm (Eu) and 665 nm (AlexaFluor 647).
MAP4K3 Biochemical Assay
The MAP4K3 biochemical assay was performed similarly to the HPK1 assay described above with the following changes. The final concentration of MAP4K3 was 1 nM and of ATP was 100 µM, or as otherwise indicated. The substrate peptide, Lance Ultra Ulight-p70 S6K (Thr389), and Lance Ultra Europium-anti-phospho-p70 S6K (Thr389) antibody were present at final concentrations of 100 and 0.125 nM, respectively. The reaction was incubated at room temperature for 2 h prior to quenching.
Compound Preparation and Dilution for Biochemical Inhibition Assays
Compounds were solvated as 10 mM stocks in DMSO and were further diluted to 1 mM or 100 µM as needed for the concentration range to be tested. Compounds were serially diluted threefold in Echo LDV plates using a Hamilton STAR Plus liquid handler and dispensed into assay plates using an Echo acoustic dispenser (Labcyte). For inhibition assays, compounds were preincubated with either HPK1 or MAP4K3 for 30 min prior to initiation by addition of ATP and peptide/protein substrate. After 60 or 120 min (HPK1 and MAP4K3, respectively) of incubation at room temperature the reactions were quenched as described above. The TR-FRET signal was normalized relative to control wells containing DMSO only or 1 µM of the known inhibitors, sunitinib malate (HPK1) and staurosporine (MAP4K3).
Human pSLP-76 ELISA
Human PBMCs were thawed, counted, and incubated in RPMI GlutaMAX supplemented with NEAA, sodium pyruvate, and 10% fetal bovine serum overnight in a 37 °C incubator with 5% CO2. The next day, cells were counted again and seeded on 384-well tissue culture plates at a density of 60,000 per well in a volume of 10 µL with columns 1 and 24 as no-cell blank controls. Intermediate plates containing 2× compounds were made by acoustic transfer (Echo; Labcyte) of 100 nL of compound and the addition of 50 µL of media, followed by a brief spin and 1 min of shaking at 1200 rpm. A Hamilton liquid dispenser was used to add 10 µL of fresh media containing 2× compounds to the cell plate, followed by a brief spin to collect liquid at the bottom of the plate. Plates were incubated in a 37 °C incubator supplemented with 5% CO2 for 1 h. T cells were activated by adding 10 µL of anti-CD3/CD28 Dynabeads (2:1 beads/cells) to the plate and 10 µL of media was used as a negative control. After a 30 s shake at 1200 rpm followed by a brief spin, plates were incubated in a 37 °C incubator with 5% CO2 for 25 min. Using the Hamilton liquid dispenser, 10 µL of 4× lysis buffer containing protease/phosphatase inhibitor was added, followed by a brief centrifugation and 10 min of shaking at 1000 rpm. Plates were immediately placed in a −80 °C freezer. They were thawed the following day on 37 °C heat blocks, followed by spinning at 3500 rpm at 4 °C for 5 min. Cell lysates (20 µL) were transferred to 384-well pSLP-76 ELISA plates and incubated for 2 h at room temperature with constant shaking at 1000 rpm. After washing plates six times with 1× wash buffer, 20 µL of detection antibody was added and incubated for 1 h at room temperature with constant shaking at 1000 rpm. After washing the plates six times with 1× wash buffer, 20 µL of horseradish peroxidase-conjugated secondary antibody was added and incubated for 1 h at room temperature with constant shaking at 1000 rpm. After washing the plates six times with 1× wash buffer, 20 µL of TMB substrate solution was added for blue color development and 20 µL of stop solution was added. Plates were read at 450 nm using media-only wells (no cells) as a reference blank.
Human IL-2 Assay
Human PBMCs were thawed, seeded, and treated with HPK1 inhibitors identically as mentioned in the pSLP-76 ELISA above. The major difference is that cells were incubated with anti-CD3/CD28 Dynabeads overnight for 16–18 h in a 37 °C incubator with 5% CO2. Supernatants taken from the cell plates were diluted eightfold with 1× PBS. Diluted supernatants (10 µL) were transferred to human IL-2 MSD ELISA plates and the assay was performed according to the manufacturer’s instructions.
Mouse pSLP-76 SiMoA Assay
To isolate mouse T cells, fresh mouse spleens were crushed in fresh T-cell media. Pan T cells were isolated using a Miltenyi Pan T-cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). T cells were seeded at the density of 100,000 cells/well on 96-well plates and were preincubated with HPK1 inhibitors for 10 min followed by stimulation with Miltenyi’s mouse anti-CD3/CD28 Dynabeads for 30 min at the 1:1 cell-to-bead ratio. Cells were then lysed and frozen at −80 °C. Anti-mouse pSLP-76 antibody was conjugated to Quanterix proprietary beads and anti-pSLP-76 antibody was biotinylated according to the manufacturer’s instructions (Quanterix Corp., Billerica, MA). Cell supernatants were transferred to an SiMoA 96-well, V-bottom plate, and the volume was made up to 100 µL using 1× lysis buffer containing protease/phosphatase inhibitor. SLP-76 capture antibody-coated beads (25 µL) were added to the cell lysates and the plate was shaken at 800 rpm at 25 °C for 30 min. An automatic SiMoA washer loaded with a three-step assay protocol was used to wash the plate. Then, 100 µL of biotinylated detection antibody was added at the final concentration of 0.3 µg/mL and the plate was shaken at 800 rpm at 25 °C for 10 min. The plate was washed again and 100 µL of streptavidin-beta-glycosidase solution was added to give a final concentration of 150 pM, and the plate was shaken at 800 rpm at 25 °C for 10 min. After final washes on the washer, the plate and fluorescent substrate solution were loaded on the SR-X instrument (Quanterix Corp.) for signal detection.
Data Analysis
Spotfire was used as a tool to perform compound curve fitting. Proper positive and negative controls were added on each plate for Z′ calculations. Dose–response curves were fitted using a four-parameter logistic fit in Spotfire and IC50 values were automatically calculated by the software (Tibco, Somerville, MA).
Results
HPK1 Biochemical Assay Development
Potent ATP-competitive inhibitors of HPK1 kinase activity have been reported by several research groups.23,24 To enable internal HPK1 inhibitor discovery efforts, a mechanistically agnostic high-throughput assay was developed. This assay needed to provide sufficient sensitivity to characterize HPK1 inhibitors with IC50 values as low as approximately 50 pM and utilize SLP-76 protein as the most physiologically relevant substrate. 1 Full-length SLP-76 with a C-terminal octa-histidine tag was produced in E. coli. To detect HPK1-mediated phosphorylation of SLP-76, a TR-FRET donor–acceptor pair consisting of an Eu-W1024-anti-6xHis antibody and an AlexaFluor 647-conjugated anti-pSLP-76 Ser376 antibody was utilized. Phosphorylation of SLP-76 increased linearly with time ( Fig. 1A ) and was dependent on HPK1 and ATP concentrations ( Fig. 1B ). The apparent KM for ATP was 8.9 ± 2.4 µM, which is consistent with the previously reported values. 25 The apparent KM for SLP-76 could not be accurately determined but is less than 2 nM (data not shown).
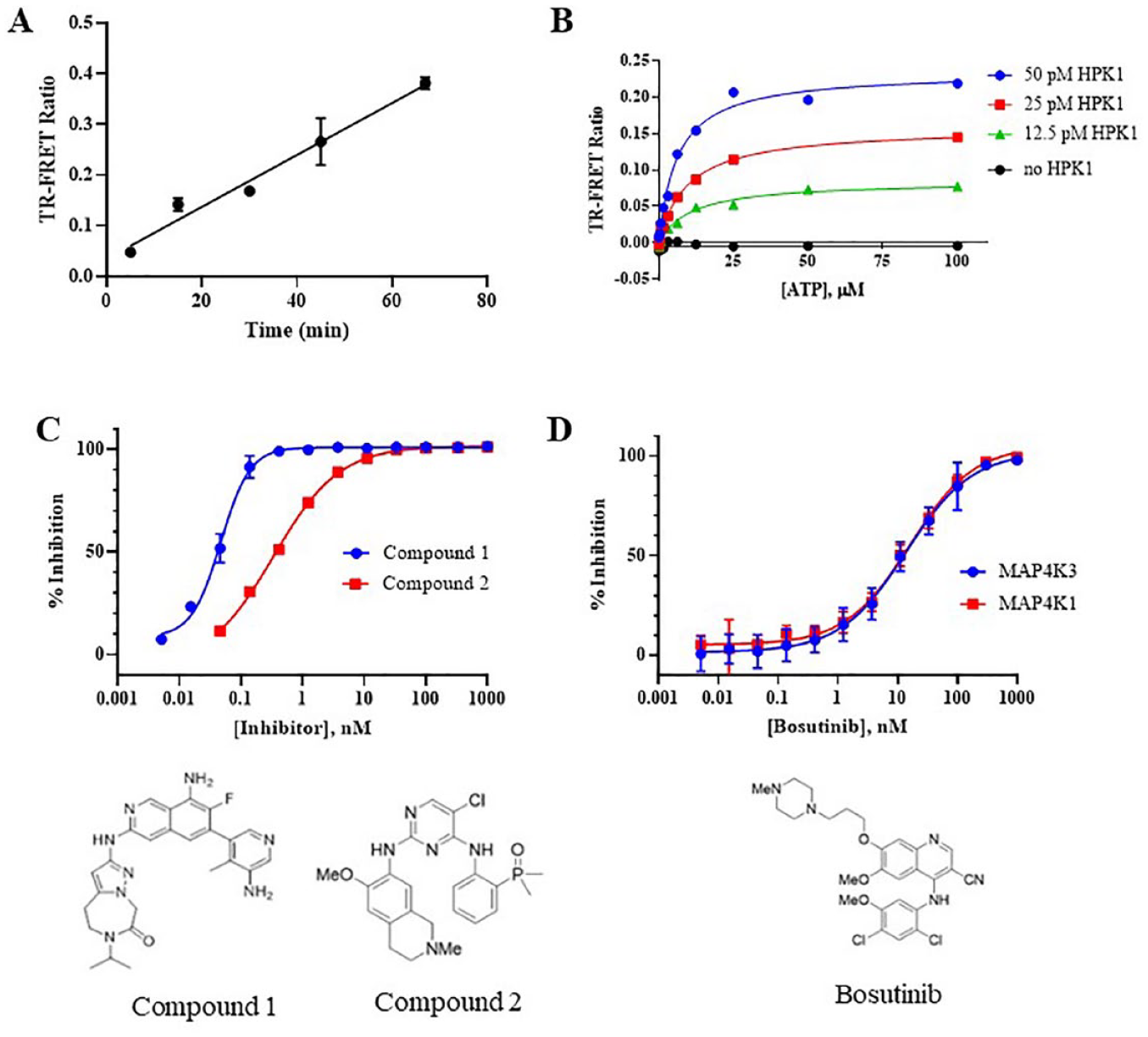
Phosphorylation of human SLP-76 as a measure of HPK1 and p70 S6K as a measure of MAP4K3 activity and inhibition by small molecules. (
To assess the selectivity of HPK1 inhibitors against the highly homologous MAP4K3 enzyme, 1 a similar TR-FRET assay was employed. MAP4K3 phosphorylates PKC-θ at Thr538; 26 however, no commercially available antibody reagents specific for this residue were identified. Therefore, a generic ULight-p70 S6K Thr389 peptide substrate was used as a substrate. In this assay, the phosphorylation of the substrate peptide increased linearly with time as a function of MAP4K3 concentration ( Suppl. Fig. S1A ). Titration of peptide at a near-saturating concentration of ATP showed the apparent KM value to be 378 ± 52 nM ( Suppl. Fig. S1B ). Due to limitations of peptide supply, the apparent KM for ATP was determined at 100 nM of peptide and was 57.3 ± 15 µM ( Suppl. Fig. S1C ). These biochemical assays were adapted to a 384-well format and used for inhibitor screening with Z′ values of routinely >0.8 and >0.6 for HPK1 and MAP4K3, respectively ( Suppl. Fig. S1D,E ). Inhibition of HPK1 by literature compounds designated as compounds 1 and 2 was determined, exhibiting IC50 values of 47 and 334 pM, respectively ( Fig. 1C ). Inhibition of HPK1 and MAP4K3 by the nonselective inhibitor bosutinib was determined and the measured IC50 values were 15.9 and 13.8 nM, respectively ( Fig. 1D ).
Development of an ELISA-Based pSLP-76 Assay Using Human PBMCs
Jurkat WT and three HPK1 null clones (clones B1, C11, and D9) were generated through CRISPR technology and used to evaluate whether phosphorylation of Ser376 of SLP-76 (pSLP-76) is a suitable readout for an HPK1 TE assay. In unstimulated Jurkat cells, no SLP-76 Ser376 signal could be detected by Western blot, whereas the total SLP-76 (tSLP-76) protein level is comparable in both WT and three HPK1-deficient Jurkat clones (
Fig. 2A
). Stimulation with anti-human CD3/CD28-antibody-coated Dynabeads for 30 min increased the pSLP-76 signal in WT but not in HPK1-deficient cells, indicating that phosphorylation of SLP-76 on Ser376 is predominantly, if not exclusively, mediated by HPK1. To develop a high-throughput assay for pSLP-76 detection, three different technologies were compared using cell lysates made from Jurkat WT cells in the absence/presence of anti-human CD3/CD28 Dynabeads. Cell lysates were serially diluted twofold five times and dilution buffer was used as a blank control. The fold change was determined by dividing the pSLP-76 signal in anti-human CD3/CD28 Dynabead-treated Jurkat WT cells by that in vehicle-treated cells. A hook effect was observed with both AlphaLISA and homogeneous time-resolved fluorescence (HTRF) technologies, and a sufficient assay window up to five- to sevenfold was only detected within a narrow range of protein concentrations (
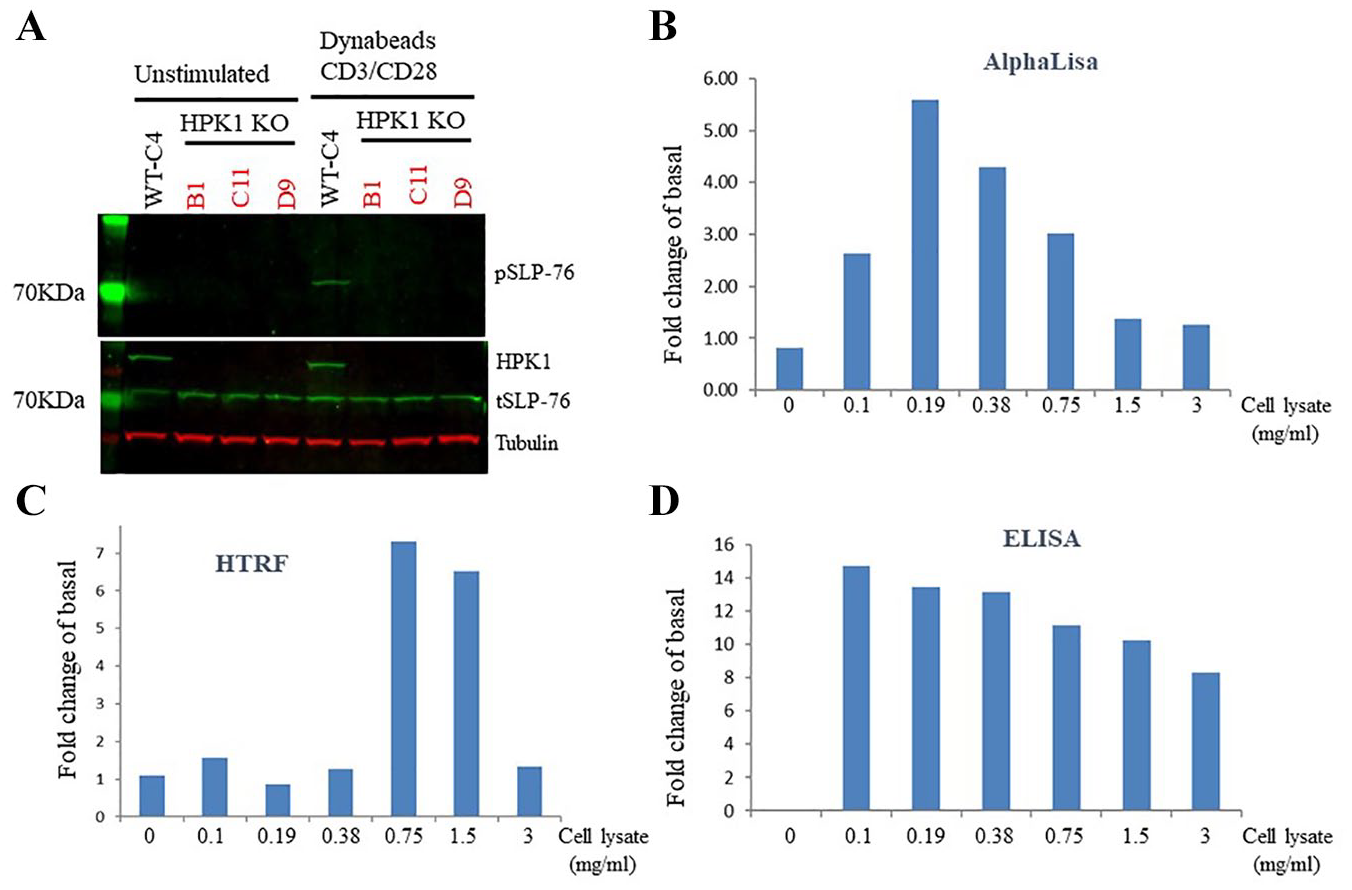
Determination of human pSLP-76 as a readout of an HPK1 TE assay. (
Although as a T-cell line Jurkat cells were useful to evaluate assay methodology, this cell line harbors millions of mutations and irregularities that have been observed in the expression of central regulators of T-cell receptor signaling pathway, 27 suggesting that this cell line may not reflect physiological responses of primary human T cells, and therefore it was not pursued further. To provide greater relevance to T-cell responses in the clinic, human PBMCs were chosen for the pSLP-76 TE assay. PBMCs from six donors were tested for the extent of SLP-76 phosphorylation in response to anti-human CD3/CD28 stimulation ( Fig. 3A ). Increased SLP-76 phosphorylation on Ser376 residue was observed in five donors with a fold change ranging from 2.7 to 6.9 ( Fig. 3B ). Donor LP_384 did not respond to this activation, suggesting a lack of HPK1 activation. Two donors, LP_353 and LP_374, showed the biggest fold change (five and seven folds over basal levels, respectively) and were chosen for the routine TE screening assay. Several cell densities and bead/cell ratios were tested in a 384-well format, and the assay condition was set at 60,000 cells/well with a bead/cell ratio of 2:1. The final conditions resulted in the assay window of 6.7 for donor LP_353 with a Z′ of 0.64, and of 9.9 for donor LP_374 with a Z′ of 0.61.
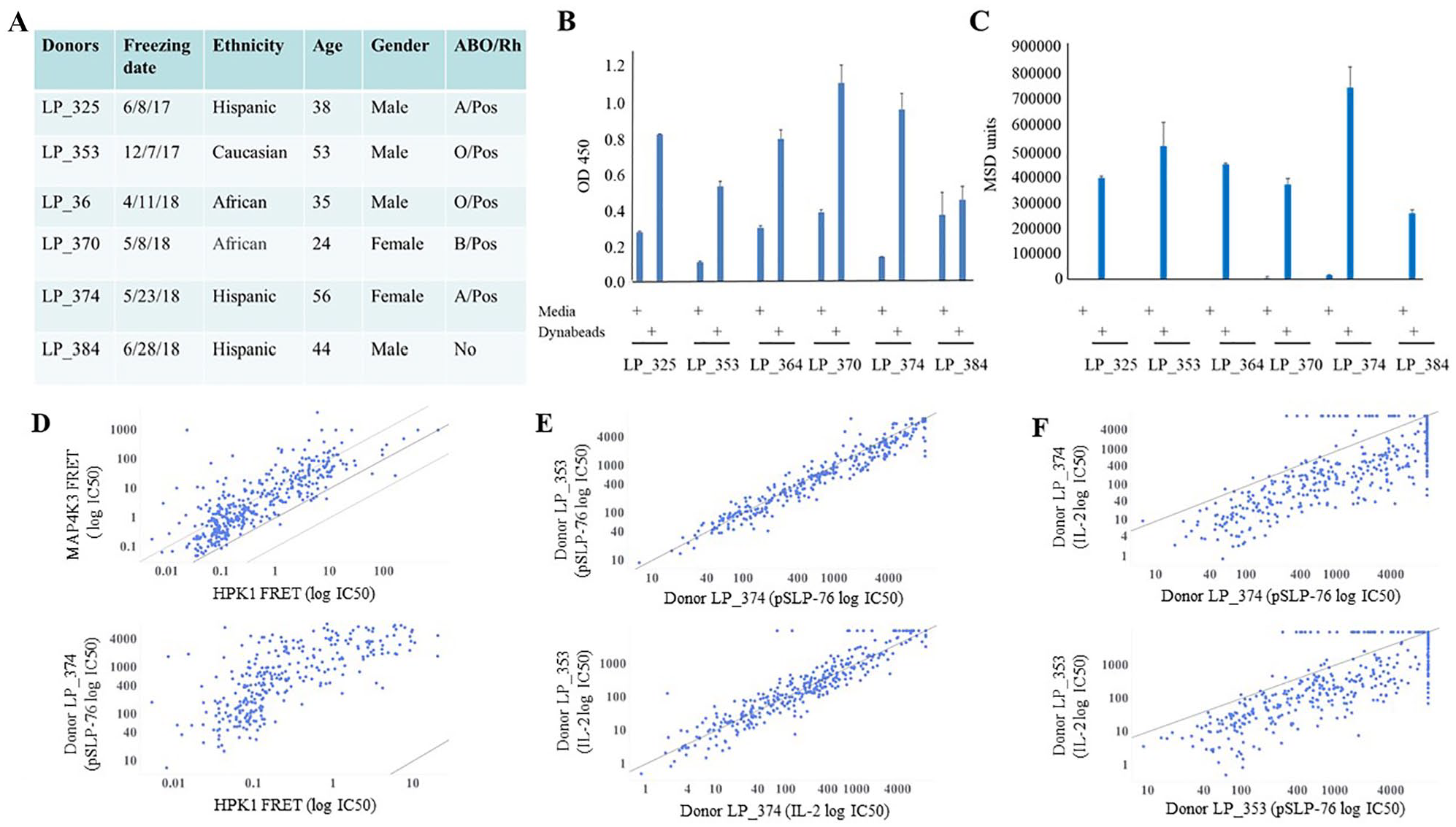
Selection of human PBMC donors and establishment of high-throughput pSLP-76 and IL-2 assays. (
Development of an MSD-ELISA-Based IL-2 Assay Using Human PBMCs
HPK1 is a negative regulator of T-cell signaling and small-molecule HPK1 inhibitors are hypothesized to enhance T-cell activities, which can be reflected by increased Th1-type cytokine secretion levels. IL-2 is secreted by both primary T cells and Jurkat cells upon activation of the T-cell receptor, which is a critical cytokine for regulating immune function. Compared with the WT and parental cells, the three HPK1-deficient Jurkat clones had distinct morphologies and varied IL-2 secretion in response to T-cell receptor stimulation despite the observation that SLP-76 Ser376 phosphorylation was consistently diminished in all of them ( Suppl. Fig. S2 ). Upon stimulation with anti-human CD3/CD28 Dynabeads, clone B1 had drastically increased IL-2 secretion, whereas clone C11 had similar levels and clone D9 had lower IL-2 secretion compared with the WT clone and parental cells. These results indicate that Jurkat cells are also not suitable for a functional assay.
To overcome this problem, the same six donors tested for the pSLP-76 assay were also used to test IL-2 secretion in response to the stimulation with anti-CD3/CD28 Dynabeads. All six donors had robust IL-2 secretion upon stimulation ( Fig. 3C ). Interestingly, the two donors with the largest pSLP-76 TE assay windows, LP_353 and LP_374, were also characterized by the highest levels of IL-2 secretion. To be consistent with the pSLP-76 assay, donors LP_353 and LP_374 were also chosen for the IL-2 assay as a distal functional readout of HPK1 activity. Optimized assay conditions were consistent with the pSLP-76 assay except 16–18 h of anti-CD3/CD28 Dynabead stimulation. A control small-molecule HPK1 inhibitor was used to define the assay window, 28 which was defined as a fold change over the DMSO vehicle control in the presence of anti-CD3/CD28 Dynabeads. The final conditions yielded assay windows of 1.8 for donor LP_353 and 2.6 for donor LP_374 with Z′ values of 0.33 and 0.49, respectively. Cell viability was determined by CellTiter-Glo reagent detection after the supernatants from treated cells were removed for IL-2 measurement.
Evaluation of HPK1 Inhibitors
To evaluate the reliability and reproducibility of pSLP-76 and IL-2 assays, a set of 31 compounds consisting of kinase inhibitors with various potencies toward HPK1 alongside inactive controls were used to treat PBMCs from donors LP_353 and LP_374. Ten-point titration curves with threefold serial dilution starting with a top concentration of 10 µM were generated. For the pSLP-76 assay, the IC50 MSR 29 was 2.51 for donor LP_353 between two independent experimental repeats ( Suppl. Table S1 ) and 2.69 between the two donors. The MSR was 1.98 for the IL-2 assay between the two donors despite the small assay window. These data indicated consistent results between experiments and good correlation between two donors.
Use of Biochemical and Cellular Assays to Support Medicinal Chemistry Efforts
Our routine assay workflow involved an initial test of all medicinal chemistry-generated compounds in the HPK1 and MAP4K3 biochemical assays. Although the nominal concentration of MAP4K3 in the assay was 1 nM, inhibitor potencies as low as 50 pM were observed with no evidence of tight-binding behavior, suggesting a high level of inactive kinase present in this preparation. Considering data from a set of 392 literature and proprietary compounds, most exhibited 3- to 100-fold selectivity for HPK1 over MAP4K3 and potencies against the two enzymes were well correlated (r2 = 0.66) ( Fig. 3D , top panel). Additional selectivity screening was performed using a panel of relevant kinase enzymatic assays (minimally consisting of MAP4K2, MAP4K4, MAP4K5, MAP4K6, JAK1, JAK3, FLT3, CDK2, RET, LCK, and STK4) performed either internally or at Life Technologies. Due to the large cell-based IC50 shift observed with HPK1 inhibitors, a cutoff on biochemical potency was employed to progress compounds into cellular assays. Only compounds with an HPK1 enzymatic IC50 of 10 nM or lower were then screened in the pSLP-76 and IL-2 assay against PBMCs from both donors. In addition to the pSLP-76 and IL-2 readouts, compounds were also tested for cytotoxicity using a bioluminescent assessment of cellular ATP levels. As shown in the bottom panel of Figure 3D , the biochemical and pSLP-76 cellular assay potencies are correlated (r2 = 0.53 for the 299 compounds with measurable cellular IC50 values against donor LP_374) but right-shifted by approximately three orders of magnitude in the cellular assay. Excellent correlations were observed for both the pSLP-76 and IL-2 assays between the two donors (r2 = 0.93 and 0.90, respectively) ( Fig. 3E ). Considering only active compounds, the correlations between the pSLP-76 and IL-2 assay were 0.65 and 0.63 for donors LP_374 and LP_353, respectively ( Fig. 3F ).
Many nonspecific HPK1 inhibitors appear to decrease phosphorylation of SLP-76 on Ser376 without inducing IL-2 secretion. Bosutinib, a small-molecule BCR-ABL and Src tyrosine kinase inhibitor, also inhibits HPK1 activity with an IC50 value of around 500–700 nM ( Fig. 4A , left panels), but this molecule actually dose-dependently decreased IL-2 secretion ( Fig. 4B , left panels). One possible explanation is that this compound can pan-inhibit Src family tyrosine kinases and therefore attenuate TCR signaling30,31 alongside cell toxicity ( Fig. 4C , left panels), which would also be expected to reduce IL-2 secretion.
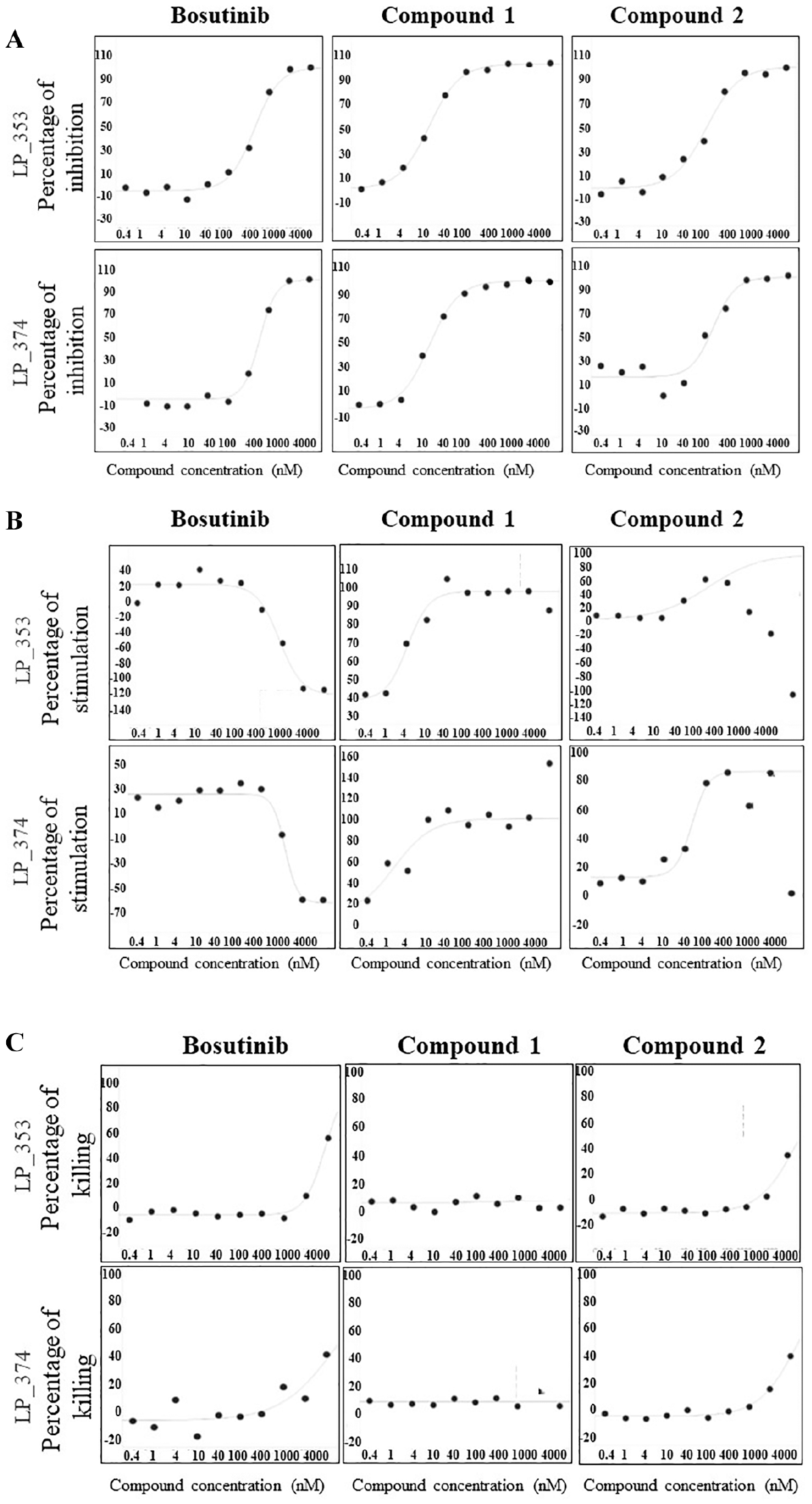
Comparison of TE versus potency of three HPK1 inhibitors using established high-throughput PBMC-based cell assays. Curves were plotted with 10-point threefold serial dilutions with a top concentration of 10,000 nM (X axis). (
To understand the utility of these biochemical and cellular assays to differentiate compounds, structure–activity relationships were explored with particular focus on two previously described series of HPK1 inhibitors. Compound 1, described by Genentech,
24
is a representative of a set of benzimidazole inhibitors referred to as series 1. A second series of aminopyrimidines from Ariad
23
(series 2) is exemplified by compound 2. Compound 1 demonstrated IC50 values of approximately 20 nM in the pSLP-76 assay and 2–5 nM in the IL-2 secretion assay (
Development of SiMoA-Based pSLP-76 Assay as a PD Biomarker for Mouse Studies
In contrast to the human PBMC pSLP-76 assay, the mouse pSLP-76 commercial ELISA kits tested had poor sensitivity, requiring about 250,000 purified T cells from mouse spleen to generate a consistent signal. These experimental conditions limited its utility as a PD marker for in vivo studies (data not shown). To improve pSLP-76 detection, a highly sensitive immunoassay technology, an SiMoA platform, was used to develop an in-house assay. A mouse SLP-76 antibody was conjugated to Quanterix proprietary beads to capture total SLP-76 protein, and a pSLP-76 Ser376 antibody was biotinylated and used as a detection reagent. The sensitivity of this assay was compared with that of a commercial mouse pSLP-76 Ser376 ELISA kit. Mouse T cells isolated from spleen were treated with either vehicle or anti-mouse CD3/CD28 Dynabeads, and cell lysates were extracted and serially diluted up to 800-fold. The SiMoA technology enabled more than a 30-fold increase in the detection of pSLP-76 signal in response to stimulation with anti-mouse CD3/CD28 Dynabeads compared with the vehicle control even after lysates were diluted 800-fold ( Fig. 5A ). In contrast, the assay window was completely lost with a commercial ELISA after cell lysates were diluted only 50-fold. The dramatically improved sensitivity with the SiMoA platform enables detection using a small amount of proteins and is suitable for PD biomarker detection in animal studies. The IC50 value for a potent HPK1 inhibitor 28 (compound 64) in mouse splenocytes is shown in Figure 5B . Compound 64 dose-dependently inhibited anti-mouse CD3-induced pSLP-76 Ser376 levels in the spleen of normal Balb/c mice ( Fig. 5C ).
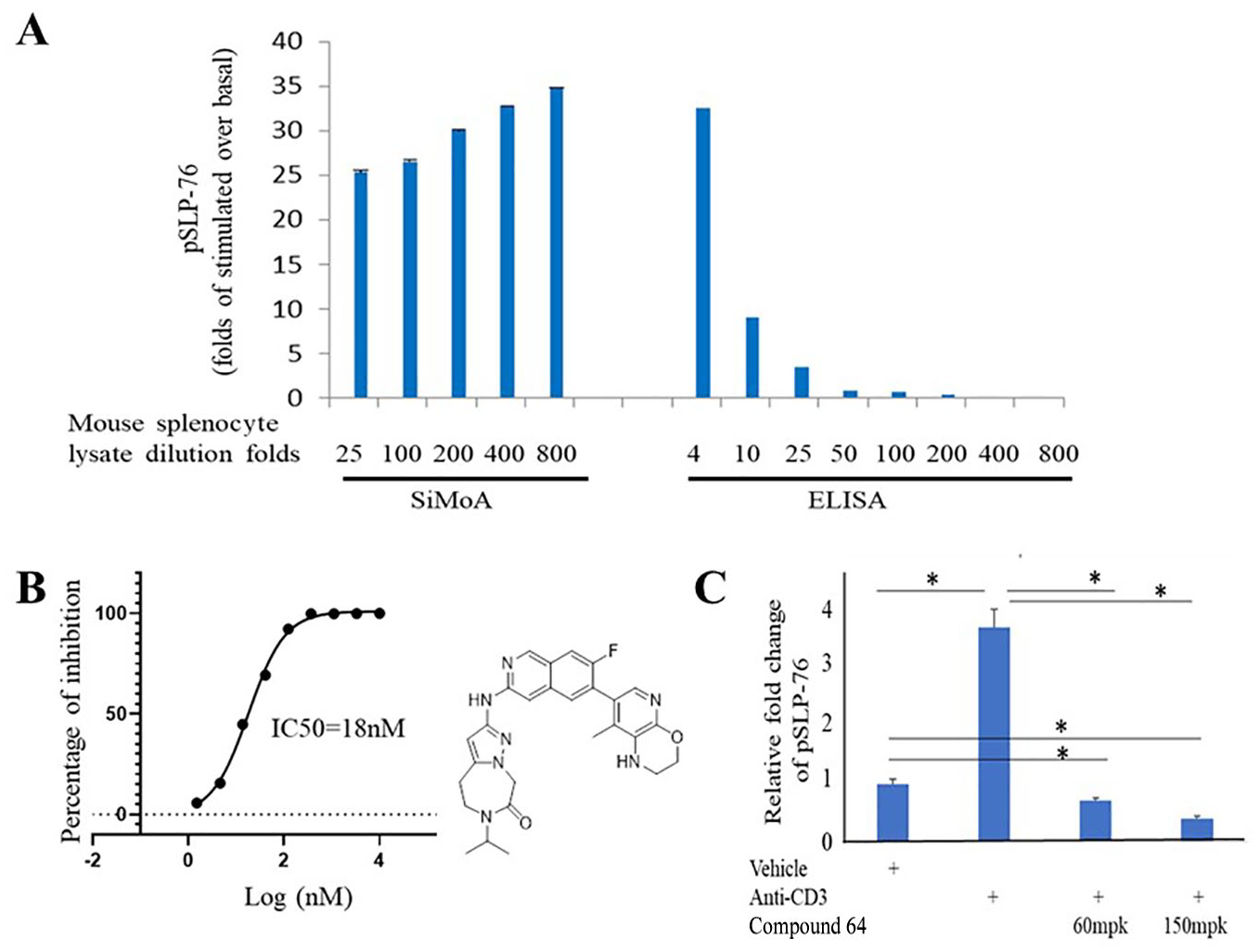
Home-brew SiMoA assay for the detection of pSLP-76 in mouse cells and tissues. (
Discussion
Cancer therapy has been evolving rapidly in the recent decade. Radiation and chemotherapies are traditional methods for treating nonsurgically removable tumors but can cause damage to healthy cells as well. 12 Targeted therapy has proven effective when the molecular etiology of the cancer type is clearly understood, with successful examples such as Gleevec for treating chronic myelogenous leukemia and Lynparza for treating breast and ovarian cancer with BRCA gene mutations.35–38 In contrast to the limited use of targeted cancer therapy, anticancer immunotherapy will train the immune system to overcome the immune-evasive feature of many cancer types. Immunotherapy has provided novel options for cancer treatment and significantly improved patient survival during the past several years, with Keytruda as the most successful example. 16 However, the majority of cancer patients have compromised immunity due to the immunosuppressive factors secreted by cancer cells.39,40 HPK1 small-molecule inhibitors could potentially further boost immune function by lifting the negative feedback mechanisms imposed on T and dendritic cells due to HPK1 activation. Among the tested six healthy donors, we observed that one donor was deficient in HPK1 activation for unknown reasons, which indicates that a subset of patients with compromised HPK1 function may not benefit from HPK1 inhibitors. In this paper, we described the establishment of various biochemical and cell-based assays to enable a drug discovery program for the identification of selective and potent HPK1 small-molecule inhibitors.
Our strategy uses an HPK1 biochemical TR-FRET assay as the tier 1 filtering assay to eliminate compounds with IC50 values of >10 nM and a 12-kinase panel to understand determinants of selectivity. HPK1 inhibitors passing selection criteria from biochemical assays were followed up with testing in two independent cell-based assays: a TE assay detecting the SLP-76 Ser376 phosphorylation level and functional assessment by measuring IL-2 secretion. Despite the fact that use of cancer cell lines is a common practice for drug discovery programs, it is advantageous to use primary T cells, which better reflect on physiological T-cell responses. Thus, the benefit of our cell-based assays is the use of PBMCs from two heathy donors instead of commonly used Jurkat cells.
Immunotherapy reagents usually first activate immune cells, which then kill the cancer cells. Unlike the tumor cells, which have a high mutation load, the immune cells from patients with solid tumors are not expected to be malignant. In contrast, Jurkat cells harbor millions of mutations and likely function very differently from primary T cells. 27 In this study, we found that three HPK1-deficient Jurkat clones had three different outcomes in terms of IL-2 secretion, despite all lacking pSLP-76 levels. These data suggest that any screening results would have been biased and unreliable had we chosen a clone with the desired phenotype and ignored the other two clones that showed unexpected profiles. It is also expected that different individuals would show variation in HPK1 activities and the extent of negative feedback in TCR signaling. To address interdonor variability, we initially screened six healthy donors and selected the same two donors for both pSLP-76 and IL-2 assays. The correlations between assays and donors are encouragingly good for active compounds defined as having IC50 values below 10 µM in cell-based assays. We also observed that some compounds active in biochemical assay are inactive in cells, which could be due to insufficient cell penetration, differences in media binding, or interference with the biochemical assay readout. Compounds that inhibit pSLP-76 but have poor selectivity usually do not enhance or even inhibit IL-2 secretion or show a bell-shaped dose–response profile. Despite these challenges, novel selective and potent HPK1 inhibitors have been identified through studying structure–activity relationships, and the evolution of these compounds will be discussed in future publications.
We further addressed the sensitivity issue of using pSLP-76 Ser376 as an in vivo PD marker in preclinical species. Unlike the human commercial pSLP-76 ELISA kit, the mouse pSLP-76 commercial ELISA kit has poor sensitivity and a consistent signal could only be detected when approximately 250,000 purified T cells were used, making it impractical for routine use. SiMoA technology is the most sensitive immunoassay reported to date, and we demonstrated that this platform is indeed at least 200-fold more sensitive than the mouse ELISA kit, even when the same pair of capture and detection antibodies was used. The successful application of SiMoA technology enabled us to monitor pSLP-76 as a PD biomarker in our mouse studies.
In summary, our extensive efforts resulted in the successful establishment of a suite of essential assays enabling the execution of our HPK1 drug discovery program and led to characterization and optimization of HPK1 inhibitors.
Supplemental Material
Lacey_supplemental_tables – Supplemental material for Development of High-Throughput Assays for Evaluation of Hematopoietic Progenitor Kinase 1 Inhibitors
Supplemental material, Lacey_supplemental_tables for Development of High-Throughput Assays for Evaluation of Hematopoietic Progenitor Kinase 1 Inhibitors by Brian M. Lacey, Zangwei Xu, Xiaomei Chai, Jason Laskey, Xavier Fradera, Payal Mittal, Sasmita Mishra, Jennifer Piesvaux, Peter Saradjian, Lynsey Shaffer, Galya Vassileva, Catherine Gerdt, Yun Wang, Heidi Ferguson, Dustin M. Smith, Jeanine Ballard, Steven Wells, Rishabh Jain, Uwe Mueller, George Addona, Ilona Kariv, Joey L. Methot, Mark Bittinger, Sheila Ranganath, Robbie Mcleod, Alexander Pasternak, J. Richard Miller and Haiyan Xu in SLAS Discovery
Supplemental Material
Lacey_Suppl_Fig._S2 – Supplemental material for Development of High-Throughput Assays for Evaluation of Hematopoietic Progenitor Kinase 1 Inhibitors
Supplemental material, Lacey_Suppl_Fig._S2 for Development of High-Throughput Assays for Evaluation of Hematopoietic Progenitor Kinase 1 Inhibitors by Brian M. Lacey, Zangwei Xu, Xiaomei Chai, Jason Laskey, Xavier Fradera, Payal Mittal, Sasmita Mishra, Jennifer Piesvaux, Peter Saradjian, Lynsey Shaffer, Galya Vassileva, Catherine Gerdt, Yun Wang, Heidi Ferguson, Dustin M. Smith, Jeanine Ballard, Steven Wells, Rishabh Jain, Uwe Mueller, George Addona, Ilona Kariv, Joey L. Methot, Mark Bittinger, Sheila Ranganath, Robbie Mcleod, Alexander Pasternak, J. Richard Miller and Haiyan Xu in SLAS Discovery
Supplemental Material
Lacey_Suppl_Fig_S1 – Supplemental material for Development of High-Throughput Assays for Evaluation of Hematopoietic Progenitor Kinase 1 Inhibitors
Supplemental material, Lacey_Suppl_Fig_S1 for Development of High-Throughput Assays for Evaluation of Hematopoietic Progenitor Kinase 1 Inhibitors by Brian M. Lacey, Zangwei Xu, Xiaomei Chai, Jason Laskey, Xavier Fradera, Payal Mittal, Sasmita Mishra, Jennifer Piesvaux, Peter Saradjian, Lynsey Shaffer, Galya Vassileva, Catherine Gerdt, Yun Wang, Heidi Ferguson, Dustin M. Smith, Jeanine Ballard, Steven Wells, Rishabh Jain, Uwe Mueller, George Addona, Ilona Kariv, Joey L. Methot, Mark Bittinger, Sheila Ranganath, Robbie Mcleod, Alexander Pasternak, J. Richard Miller and Haiyan Xu in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, and may own stock or stock options in Merck & Co., Inc., Kenilworth, NJ.
Funding
The authors received no grant support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.