
Abstract
Human health is at risk from environmental exposures to a wide range of chemical toxicants and endocrine-disrupting chemicals (EDCs). As part of understanding this risk, the U.S. Environmental Protection Agency (EPA) has been pursuing new high-throughput in vitro assays and computational models to characterize EDCs. EPA models have incorporated our high-content analysis–based green fluorescent protein estrogen receptor (GFP-ER): PRL-HeLa assay, which allows direct visualization of ER binding to DNA regulatory elements. Here, we characterize a modified functional assay based on the stable expression of a chimeric androgen receptor (ARER), wherein a region containing the native AR DNA-binding domain (DBD) was replaced with the ERα DBD (amino acids 183–254). We demonstrate that the AR agonist dihydrotestosterone induces GFP-ARER nuclear translocation, PRL promoter binding, and transcriptional activity at physiologically relevant concentrations (<1 nM). In contrast, the AR antagonist bicalutamide induces only nuclear translocation of the GFP-ARER receptor (at μM concentrations). Estradiol also fails to induce visible chromatin binding, indicating androgen specificity. In a screen of reference chemicals from the EPA and the Agency for Toxic Substances and Disease Registry, the GFP-ARER cell model identified and mechanistically grouped activity by known (anti-)androgens based on the ability to induce nuclear translocation and/or chromatin binding. Finally, the cell model was used to identify potential (anti-)androgens in environmental samples in collaboration with the Houston Ship Channel/Galveston Bay Texas A&M University EPA Superfund Research Program. Based on these data, the chromatin-binding, in vitro assay–based GFP-ARER model represents a selective tool for rapidly identifying androgenic activity associated with drugs, chemicals, and environmental samples.
Keywords
Introduction
The androgen receptor (AR) is a nuclear receptor (NR), a family of 48 transcription factors in humans that regulate gene expression in response to circulating ligands. In response to endogenous androgens, the AR regulates a gene network important in multiple physiological responses, including those that result in the differentiation and maintenance of the male sexual phenotype. In common with other NRs, AR has a modular domain structure comprising a relatively large N-terminal activation function (AF-1) domain, a DNA-binding domain (DBD), and a C-terminal ligand binding domain that also harbors a ligand-dependent transcriptional activation function (AF-2). In the absence of ligand, AR usually resides in the cytoplasm in complex with chaperone proteins. Androgen binding results in a conformational change of AR, chaperone shedding, and translocation into the nucleus. Once in the nucleus, the receptor interacts with DNA androgen response elements (AREs) and protein coactivators to initiate gene transcription. 1 This is a carefully regulated process, and disruptions can result in proliferative, reproductive, and metabolic disorders, including birth defects, hormone-dependent cancers, and infertility. 1,2
Endocrine-disrupting chemicals (EDCs) are exogenous chemicals that interfere with normal hormone action. 2,3 Although the impact on estrogen receptor signaling has received the most focus, EDCs have been shown to likely affect human health through multiple NRs, including the estrogen-related receptor, pregnane X receptor, thyroid hormone receptors, and AR. 2 -4 Societal concerns about the impact of EDC exposures is reflected in governmental regulations and international guidelines that support research seeking to understand potential exposure, the effects of exposure, and how to minimize future exposure risks. 2,5 -9 These regulations and guidelines often described a tiered approach to understanding EDC activity using a combination of in vitro– and in vivo–based assays. 10,11 More recently, there has been an increased focus on deploying high-throughput in vitro screening approaches to better capture the diverse landscape of chemicals currently in use and allow the development of computational models to better predict in vivo EDC activity. 12,13
A considerable number of chemicals components, including pesticides, flame retardants, packaging chemicals, and industrial chemicals, have been identified as androgenic or anti-androgenic. 14 -16 Although in vivo assays (rat pubertal, Hershberger assay) remain the gold standard for determining androgenic activity, multiple in vitro cell models for detecting effects on AR transcriptional activity have been developed. 3,5,10,11 These reporter systems generally consist of either rodent (CHO) or human (22RV1, HeLa, MDA-MB-453, PC3, U2OS) cell lines engineered to express human AR and containing an MMTV/ARE-luciferase reporter. These cell models have been used extensively to characterize EDC activity; however, they are vulnerable to poor selectively because of endogenous progesterone, mineralocorticoid, and glucocorticoid receptor expression, NRs known to bind and activate the MMTV/ARE-luciferase reporter gene. 3,11
To overcome the potential limitations association with reporter cell models, we have used a novel cell model that has been developed based on direct visualization of NR-DNA binding, coregulator accumulation, and transcriptional activity. The estrogen-responsive biosensor cell model (GFP-ERα: PRL-HeLa) is based on a multicopy integration of prolactin promoter/enhancer elements, green fluorescent protein (GFP) fusion technology, and automated imaging and analysis. 17,18 This cell model has been used to characterize mechanisms affecting ERα and ERβ activity, including ligand specificity, receptor mobility on promoter elements, functional importance of receptor domains, and regulation by ubiquitin ligase. 18 -21 The systems-biology nature of the cell model has also allowed it to be used as a tool to characterize the EDC potential of bisphenol analogs and BPA alternatives. It is important to note that the actions of the BPA alternatives, which were almost all active, were quantified and classified by the robust mechanistic/phenotypic high-content analysis data that are derived from the PRL-Hela models. 22,23
Recently, the modular nature of NRs that allows the generation of functional chimeras has been exploited to allow the use of the PRL-HeLa cell model to understand protein kinase regulation of the progesterone receptor (PR). 24 -28 The GFP-PRER: PRL-HeLa cell model, in which the PR DBD has been swapped for the ERα DBD, demonstrated ligand specificity for progestins and anti-progestins and allowed the direct visualization of protein kinase effects on PR nuclear translocation, DNA binding, coactivator recruitment, chromatin remodeling, and transcriptional activity. 28 Here, we report the generation of a stable PRL-HeLa cell model that expresses a functional chimeric AR (ARER) based on standard in vitro transcription assays possessing the ERα DBD inserted in place of the native AR DBD. We demonstrate that the new cell model possesses quantifiable mechanistic steps associated with androgen-induced transcriptional activity and altered mechanisms associated with androgenic/anti-androgenic exposures. The speed, sensitivity, and multiple mechanistic readouts of the new model were tested for ligand specificity via the use of a focused library of EDC reference chemicals. Evaluation of this approach was performed in the context of real-life environmental samples collected in conjunction with a US EPA Superfund Research Program involving water samples collected from Galveston Bay and the Houston Ship Channel. Based on our observations, the GFP-ARER: PRL-HeLa cell model allows the rapid detection of androgenic/anti-androgenic EDC activity with potentially distinct mechanistic phenotypes.
Materials and Methods
Materials
Cell culture tetracycline-free fetal bovine serum (FBS) was obtained from Gemini Bio-products (Sacramento, CA); trypsin-EDTA was obtained from Sigma (St. Louis, MO); cell culture media, G418, hygromycin, and L-glutamine were obtained from Corning Cellgro (Corning, NY); and blasticidin was obtained from Invitrogen (Carlsbad, CA). Dihydrotestosterone (DHT), bicalutamide, and 17β-estradiol (E2) were obtained from Sigma. A set of 43 estrogenic EDC reference chemicals with well documented in vivo activity was provided in DMSO by agreement from Dr. Keith Houck (US Environmental Protection Agency [EPA], Research Triangle Park, NC). Agency for Toxic Substances and Disease Registry (ATSDR) reference chemicals and 16 complex mixtures were provided in DMSO by the lab of Dr. Ivan Rusyn (Texas A&M, College Station, TX) as part of the Superfund Research Program. Further information about chemicals used can be found in
As part of the Superfund Research Program collaboration (principal investigator, Ivan Rusyn), environmental samples (sediments) were collected by the lab of Dr. Anthony Knap (Texas A&M) and extracted by the Rusyn lab using previously established protocols. 29,30 Specifically, 1 g of freeze-dried sample was added to a 15 mL conical-bottom disposable plastic tube (Corning, Vernon Hills, IL) and mixed with 2 mL of cyclohexane (high-performance liquid chromatography grade; Fisher Scientific, Waltham, MA) and 2 mL of DMSO (≥99%, Santa Cruz Biotechnology, Santa Cruz, CA) pre-equilibrated for 24 h with cyclohexane at a 10:1 ratio. Tubes were vortexed for 1 min and centrifuged for 5 min at 4700 rpm. Next, 1 mL of DMSO layer was removed into a clean glass vial (Lab Products, Inc., Houston, TX). An additional 2 mL of DMSO was added to the tube with the sample, and vortexing/centrifugation steps were repeated. The DMSO layer (2 mL) was removed and combined with the DMSO fraction from the first step of extraction. Samples were labeled by core (1, 2, 3, etc.); where multiple samples were extracted from a single core corresponding to differing sediment depths, the sample label contained an additional alpha code (a, b, c, etc.).
Cell Model Generation
A pSG5-flag backbone vector with human AR N-terminal domain (1-E540), ERα DBD (S178-K252), and AR ligand binding domain (G627-919) were used to create and functionally test the chimeric receptor in standard transcription assays in vitro (data not shown). DNA binding to EREs was confirmed in electrophoretic mobility shift assays (data not shown). The ARER coding region was subcloned using XmaI and BamHI sites into an enhanced GFP (eGFP) expression vector, pEGFP-C1-AR 108, that contains a truncated version of full-length AR, placing the GFP tag on the N-terminus of the chimeric protein. Next, using HindIII & BamHI sites, the ARER coding region was cloned into pENTR-EGFP-C1, a vector we prepared from pENTR (Invitrogen) that contains the eGFP and C1 reading frame multiple-cloning site from pEGFP-C1. Using the recombinase method (LR Clonase II, Invitrogen), the EGFP-C1 ARER coding region was cloned into pINDUCER20, containing a tetracycline-inducible promoter, a gift from Dr. Trey Westbrook (Baylor College of Medicine, Houston, TX). High-titer lentivirus was packaged by System Biosciences LLC (Palo Alto, CA) despite eGFP-ARER exceeding the recommended packaging size. The resulting lentivirus particles were used to transduce the PRL-HeLa cell line. PRL-HeLa cells stably expressing GFP-ARER were enriched using geneticin (G418) drug selection, flow cytometry, and single-cell cloning using limiting dilution. The final population of cells was >95% GFP positive following doxycycline induction. The GFP-ARER: PRL-HeLa cell line was maintained in phenol red-free Dulbecco’s modified Eagle’s medium (DMEM-HG) supplemented with 5% FBS, 200 μg/mL hygromycin, and 400 μg/mL G418.
Cell Culture
GFP-ER: PRL-HeLa cells were maintained in phenol red-free DMEM containing 5% FBS, 0.8 μg/mL blasticidin, 200 μg/mL hygromycin, and 10 nM 4-hydroxytamoxifen as previously described. 14,18,19 GFP-ARER and GFP-ER PRL-HeLa cells were seeded in DMEM with 5% charcoal-stripped/dialyzed FBS without selection agents or inhibitors on 96-well (Greiner SensoplatePlus) or 384-well plates (384-IQ, Aurora Biotechnologies, Poway, CA) at a target density of 8000 cells/well (96) or 3000 cells/well (384). Cells were allowed to adhere overnight, and then the GFP-ARER cells were treated with doxycycline (0.8 μg/mL) for 18 to 20 h before media were exchanged prior to treatment.
C4-2 and 22RV1 cells were genotype-verified cell lines obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained according to recommended conditions in DMEM/F12 media with 10% FBS. Cells were seeded on multiwell plates in phenol red-free DMEM with 5% SD-FBS 24 h prior to use.
Treatment with Chemicals, Mixtures, and Environmental Samples
The EPA and ATSDR reference chemicals and mixtures were provided in preformatted multiwell plates in DMSO at a known concentration. In vitro screening in GFP-ARER: PRL-HeLa cells was performed at concentrations ranging from 10 nM to 10 μM (EPA), 0.1 nM to 10 μM (ATSDR), and 200-fold to 200,000-fold dilution (mixtures, samples) and a treatment time of 2 h. For EPA chemicals and environmental samples, stock chemical solutions were directly transferred to assay plates using an acoustic transfer device (Labcyte Echo 550). For ATSDR chemicals and mixtures, immediately prior to compound treatment, the stock compound solutions were transferred to 96-well working dilution plates in phenol red-free DMEM with 5% SD-FBS. Working dilutions of compounds were then added in quadruplicate to assay plates containing prepared cells. For all experiments, negative control wells containing 0.5% DMSO and positive control wells containing DHT ranging from 0.01 nM to 1000 nM were included in the plate layout.
Immunofluorescence Labeling
After the incubation period was completed, cells were fixed using 4% EM-grade formaldehyde in PEM buffer (80 mM potassium PIPES [pH 6.8], 5 mM EGTA, and 2 mM MgCl2) and quenched with 0.1 M ammonium chloride for 10 min. Cell membranes were disrupted by incubating samples with a 0.5% Triton X-100 solution for 10 min. Nuclei were stained using DAPI (1 μg/mL) for 10 min. For samples in which antibody labeling was used, incubation with 0.5% Triton-X was extended to 30 min, and cells were incubated in blotto (5% milk in Tris-buffered saline/Tween 20) for 30 min prior to addition of primary antibody solution (AR 441, 1 μg/mL, gift from Dr. Dean Edwards; SRC-1, 0.25 μg/mL, BD Transduction Labs [Franklin Lakes, NJ]; SRC-3, 1 μg/mL, gift from Dr. Bert O’Malley; BRG1, 0.9 μg/mL Bethyl [Montgomery, TX]; Ser5-phospho RNA polymerase II, 0.5 µg/mL, Abcam [Cambridge, UK]). Primary antibody solution was incubated overnight at 4 °C prior to incubation with secondary antibody (Alexa 647 conjugated anti-Ms IgG; Molecular Probes, Eugene, OR) for 1 h at room temperature. Nuclei were stained using DAPI (1 μg/mL) for 10 min. All samples were stored in phosphate-buffered saline (PBS) containing calcium, magnesium, and sodium azide at 4 °C prior to imaging.
Fluorescence In Situ Hybridization
GFP-ARER: PRL-HeLa were fixed in 4% formaldehyde in ribonuclease-free PBS for 30 min, washed in PBS, and then permeabilized with 70% ethanol in ribonuclease-free water for a minimum of 1 h at 4 °C. Cells were washed in 1 mL of wash buffer A (LGC Biosearch Technologies, Novato, CA) containing 10% v/v formamide followed by hybridization with custom-made dsRED2 RNA probes 18,19,24 (Stellaris probes; LGC Biosearch Technologies) diluted 1:500 (50 nM) in hybridization buffer (1 g dextran sulfate, 1 mL 20× saline sodium citrate buffer and 1 mL formamide in 8 mL of nuclease-free water) overnight at 37 °C. After hybridization, cells were washed twice (15 min at 37 °C) with wash buffer A and then with wash buffer B (LGC Biosearch Technologies) for 30 min at 37 °C and then stained with DAPI for 10 min at 37 °C. After DAPI labeling, the cells were washed 1× with Dulbecco’s PBS and then stored and imaged in Dulbecco’s PBS + 0.02% sodium azide.
Imaging and Quantitation
Automated imaging was carried out using either a Vala Sciences IC-200 (San Diego, CA) or a Molecular Devices ImageXpress Micro (Downingtown, PA) image cytometer. Image acquisition was performed with a 20×/0.75NA objective and a scientific CMOS camera. Z-stacks were imaged at 1 µm intervals (for a total of 5 μm) at 1 × 1 binning. Nuclear array segmentation and automated image analyses were performed using custom measurement and reporting routines incorporated into myImageAnalysis/Pipeline Pilot software. 31 Cell, nucleus, and array (if present) were identified and metrics describing shape and intensity features quantified. Aggregated cells, mitotic cells, and apoptotic cells were removed using filters based on nuclear size, nuclear shape, and nuclear intensity. The degree of nuclear translocation of the GFP signal was determined by measuring the ratio of the nuclear mean intensity to the cytoplasmic mean intensity. Loading on the PRL array was determined by calculating the ratio of the array mean intensity to the nuclear mean intensity. GFP expression per cell was determined by integrating the total GFP pixel intensity per cell.
Data Analysis
The chemical responses were analyzed using a standardized analysis pipeline generated using Pipeline Pilot (Biovia, San Diego, CA). This pipeline performed all necessary baseline correction, normalization, curve fitting, and hit calling. All screening data were normalized to a fractional range with DMSO and 100 nM DHT-treated controls set at 0 and 1. All dose-response data were fit to either a constant model or a hill model using least-squares curve fitting. The activity observed for each chemical on measured endpoints was summarized by integrating the response over the tested concentration range (area under curve [AUC]). The AUC values were normalized to DHT and labeled as significant if greater than 0.1. This threshold was selected based on EPA computational models. 26
Statistical Analysis
Data presented were acquired from a minimum of three independent experiments. Statistical significance was determined using one-way analysis of variance with Tukey’s post hoc test for comparisons across multiple samples. Tests were performed using Minitab 19 software, and p < 0.05 was considered statistically significant. All graphs were generated using SigmaPlot 10 software.
Results
Regulation of AR activity is a complex process involving expression, protein interactions, and subcellular compartmentation of the receptor. Direct analysis of AR transcriptional activity using reporter assays is routinely complicated by bulk cell analysis and the coexpression of other NRs that may generate nonspecific results; furthermore, single endpoint assays may not capture global effects on receptor activity. 11,26 To address these issues, we mirrored our previous ER studies to establish an androgenic cell model capable of measuring multiple mechanistic steps of AR signaling by high-throughput microscopy and high-content image analysis. 28 As described previously, the PRL-HeLa parental cell line contains a multicopy (∼100 copies) integration of a cassette derived from the prolactin promoter and multimerized synergy element (repeats containing a Pit-1 and ERE binding sites; see the Methods section) linked to a dsRED2 fluorescent protein reporter. 17 When GFP-ERα (or ERβ) is expressed, agonist treatment results in receptor binding at the ERE-rich locus ("promoter array"), forming a visible intranuclear GFP spot. Binding at the promoter array is associated with coregulator recruitment (visualized using immunofluorescence) and de novo transcription of the dsRED2 reporter (detected using mRNA fluorescence in situ hybridization [FISH]). 18,19,22,23 The versatility of the systems is well-exemplified by the ability to use chimeric receptors in single-cell assays within the context of additional endocrine models, as described in our PR/estrogen receptor chimera model. 28
To enable the study of AR in the PRL-HeLa cell line, a chimeric AR was generated by swapping the DBD sequence of AR with the sequence containing the DBD from ERα (e.g., ARER;
Fig. 1A
). The chimeric protein also includes a GFP protein fusion attached to the N-terminus (GFP-ARER). GFP-ARER was next cloned into a vector that included a tetracycline-inducible promoter. After stable transfection into the PRL-HeLa cell line, single-cell cloning was used to generate the final GFP-ARER: PRL-HeLa cell line that allows for culture maintenance in the absence of AR expression. Without doxycycline induction, GFP-ARER expression is negligible, and background levels of dsRED2 mRNA are observed (
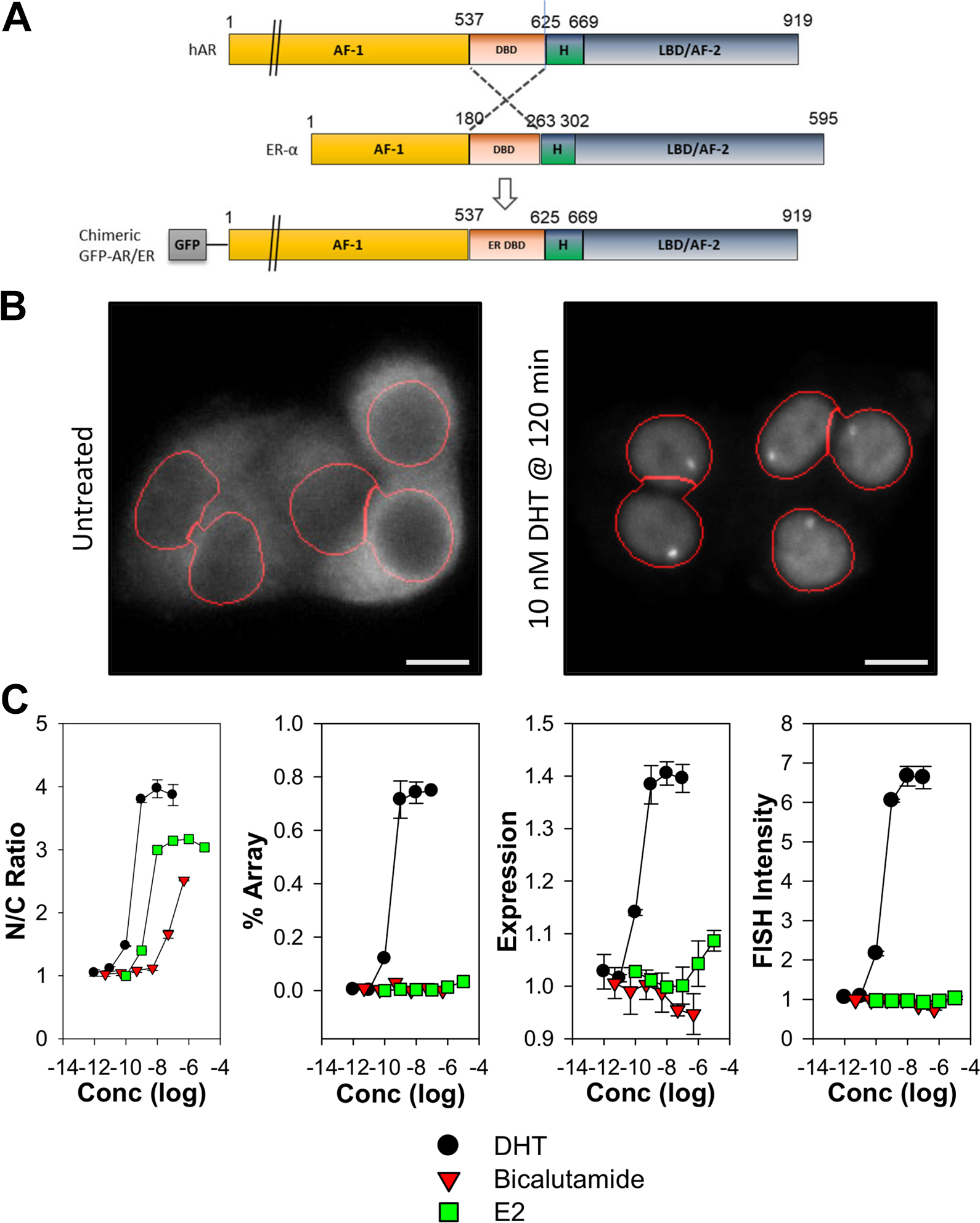
Generation and characterization of stable cell lines expressing inducible chimeric androgen receptor (AR). (
To further explore GFP-ARER ligand-dependent responses, GFP-ARER: PRL-HeLa cells were treated with increasing concentrations of DHT, the anti-androgen bicalutamide (i.e., Casodex), or 17β-estradiol (E2) and analyzed by high-content analysis (
Fig. 1B
). DHT treatment results in a dose-dependent nuclear translocation, visible array/chromatin binding, stabilization of receptor expression, and dsRED2 mRNA synthesis. These responses occurred with EC50 values ranging between 0.15 and 0.24 nM (
Table 1
), concentrations consistent with known wild-type AR sensitivity to DHT, and previously observed results using GFP-AR.
32
-34
In contrast, bicalutamide induced only nuclear translocation of GFP-ARER at an EC50 concentration greater than 200 nM (
Fig. 1C
;
Measured EC50 Values of Observed Responses.
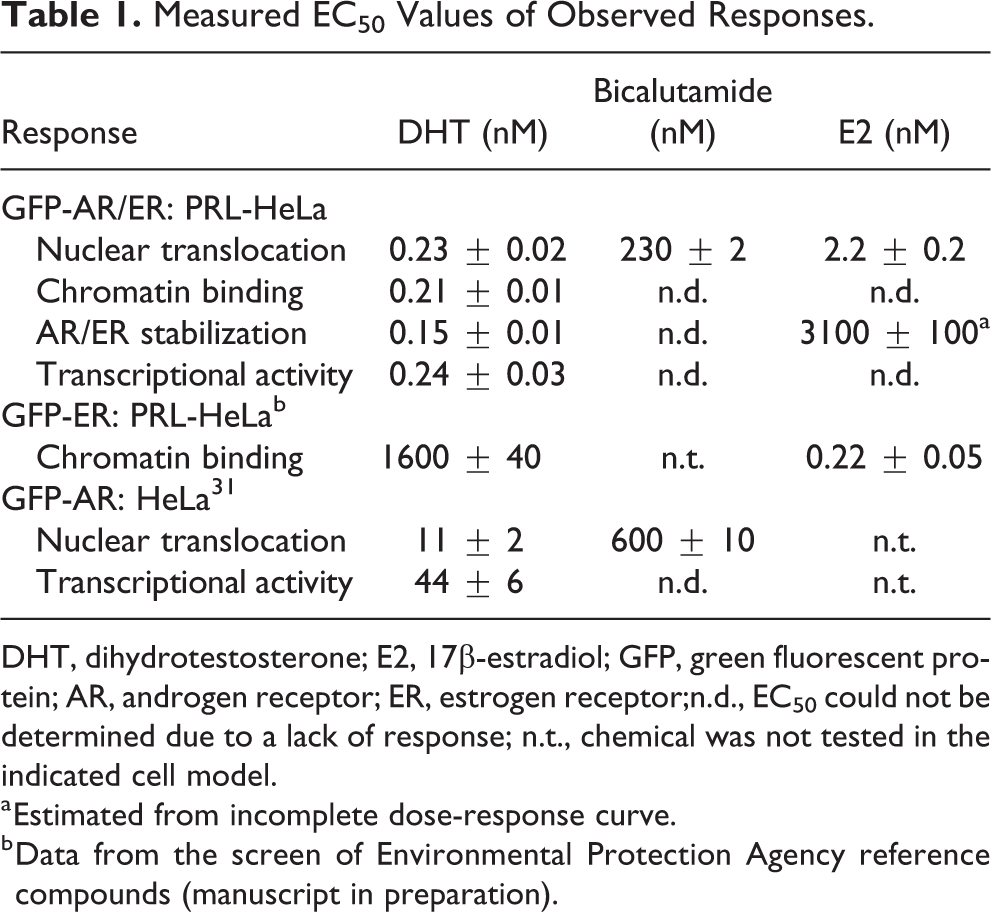
DHT, dihydrotestosterone; E2, 17β-estradiol; GFP, green fluorescent protein; AR, androgen receptor; ER, estrogen receptor;n.d., EC50 could not be determined due to a lack of response; n.t., chemical was not tested in the indicated cell model.
a Estimated from incomplete dose-response curve.
b Data from the screen of Environmental Protection Agency reference compounds (manuscript in preparation).
To understand the molecular mechanisms associated with GFP-ARER–mediated transcriptional activity, we used”‘visual ChIP” to measure coregulator protein recruitment to the integrated PRL array in the presence of DHT. Specifically, we used immunofluorescence to measure recruitment of the p160 coactivators SRC-1 and SRC-3, the chromatin remodeler BRG1, and activated RNA Pol II (serine 5 phosphorylated, S5P) in the GFP-ARER cell model compared with the GFP-ER cell model ( Fig. 2 ). We have previously used relative loading, defined as an average array signal divided by average nuclear signal, to quantify coregulator recruitment in PRL-HeLa cell lines. 18,22,23,28 In the presence of DHT, GFP-ARER cells recruited all proteins to the PRL array; however, the relative loading (array signal intensity relative to nuclear signal intensity) of the p160 coactivators SRC-1 and SRC-3 was significantly lower compared with E2-induced recruitment in GFP-ER cells despite similar recruitment of BRG1 and S5P ( Fig. 2A ). Further investigation found that this is likely a function of GFP-ARER cells having higher nuclear signal intensity of SRC-1 and SRC-3 as compared with GFP-ER cells ( Fig. 2B ). This indicates the GFP-ARER chimera can recruit SRC-1 and SRC-3 to the PRL array in a similar manner as GFP-ER, consistent with known AR interactions with the p160 coactivators and components of the transcriptional machinery. 35
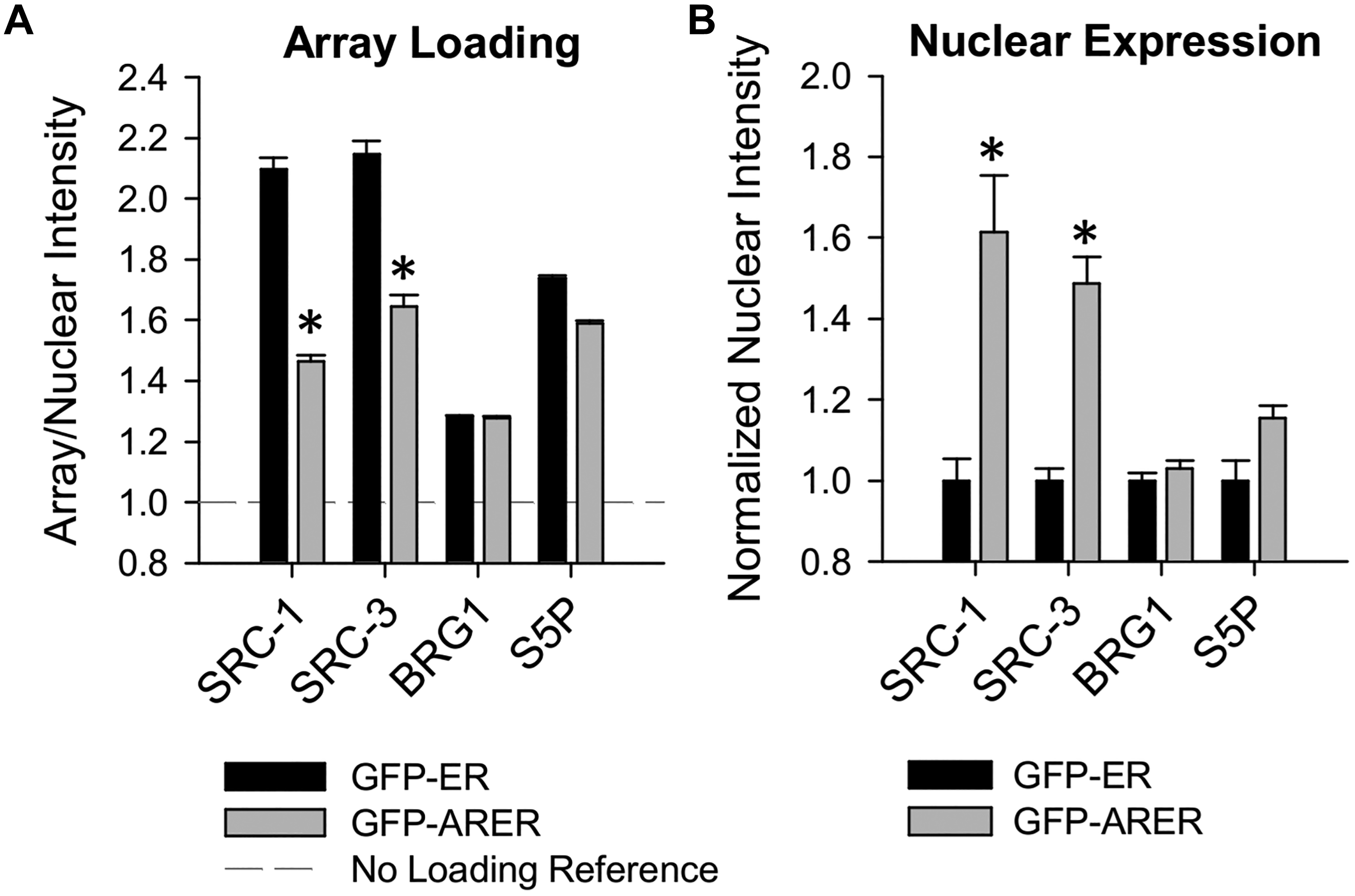
Quantification of agonist-induced coregulator and transcriptional machinery recruitment to the PRL array. GFP-ER: PRL-HeLa and GFP-ARER: PRL-HeLa cells were prepared and treated with either 10 nM E2 (GFP-ER) or 10 nM dihydrotestosterone (GFP-ARER) for 120 min. Cells were processed for immunofluorescence with antibodies against SRC-1, SRC-2, BRG1, and phosphorylated RNA Pol II (serine 5). (
Assessment of GFP-ARER: PRL-HeLa detection of androgenic bioactivity relied on chemical libraries defined by EPA or the ATSDR. The EPA reference chemicals consists of 43 chemicals with defined in vivo activity; the ATSDR reference chemicals consists of 42 pesticides, herbicides, heavy metals, or otherwise hazardous contaminants found in the environment. ATSDR individual chemicals were also combined into defined complex mixtures to mimic the complexity of environmental pollution. Leaning further toward environmental situations, we sought to also explore responses of the GFP-ARER model to endocrine disruption–relevant bisphenol A derivatives (BPXs) that have previously been exhaustively characterized by high-content analysis using the GFP-ER: PRL-HeLa cell model.
22
Further information about the chemicals and mixtures used can be found in
Within the EPA reference set of chemicals, we detected 19 that induced nuclear translocation of GFP-ARER ( Fig. 3 ). Among the strongest responders were known androgens (5alpha-DHT, 17-methyltestosterone), anti-androgens (hydroxyflutamide, promycidone), and strong agonists for other NRs (estradiols, estrone, progesterone, spironolactone, corticosterone). However, only 6 of the 19 chemicals were able to induce a visible spot indicating chromatin binding by GFP-ARER. In addition to the known androgens, these included spironolactone, estrone, 17alpha-estradiol, and 17beta-estradiol, compounds with reported weak androgenic activity. 36,37 However, the AUC of chromatin binding of these other chemicals was lower than that for DHT and 17-methyltestosterone, indicating that induction of chromatin binding required higher concentrations.
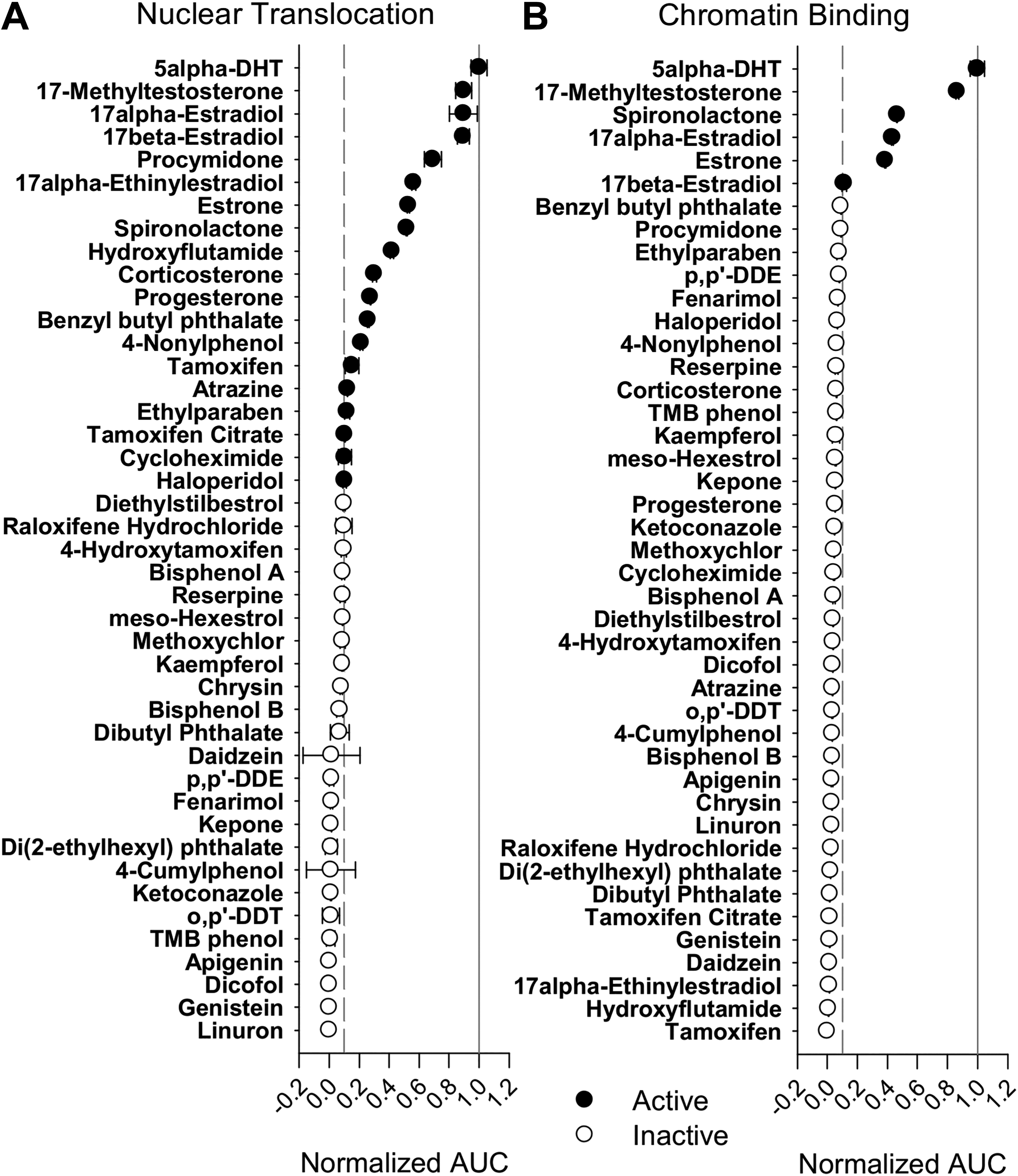
GFP-ARER as a biosensor to query for EPA reference chemicals. GFP-ARER: PRL-HeLa cells were seeded onto 384-well plates and induced with 200 ng/mL doxycycline for 24 h prior to treatment with 10 nM to 10 μM of dihydrotestosterone (DHT) or 43 reference chemicals for 2 h. Calculated area under the curve scores for (
The performance of the supplemented ATSDR chemicals and mixtures further confirmed the selective nature of GFP-ARER chromatin binding (
To understand the relationships between the observed responses, we compiled nuclear translocation, chromatin binding, and receptor stabilization metric AUC data from active chemicals/mixtures. All data were standardized to a range based on vehicle and DHT controls and clustered using a k-means algorithm (
Fig. 4
). Cluster 1 contains chemicals that induced a high AUC score for nuclear translocation, chromatin binding, and receptor stabilization. It contains 5alpha-dihydrotesterone and 17-methyltestosterone, the only two strong/moderate AR agonists in the chemicals tested (
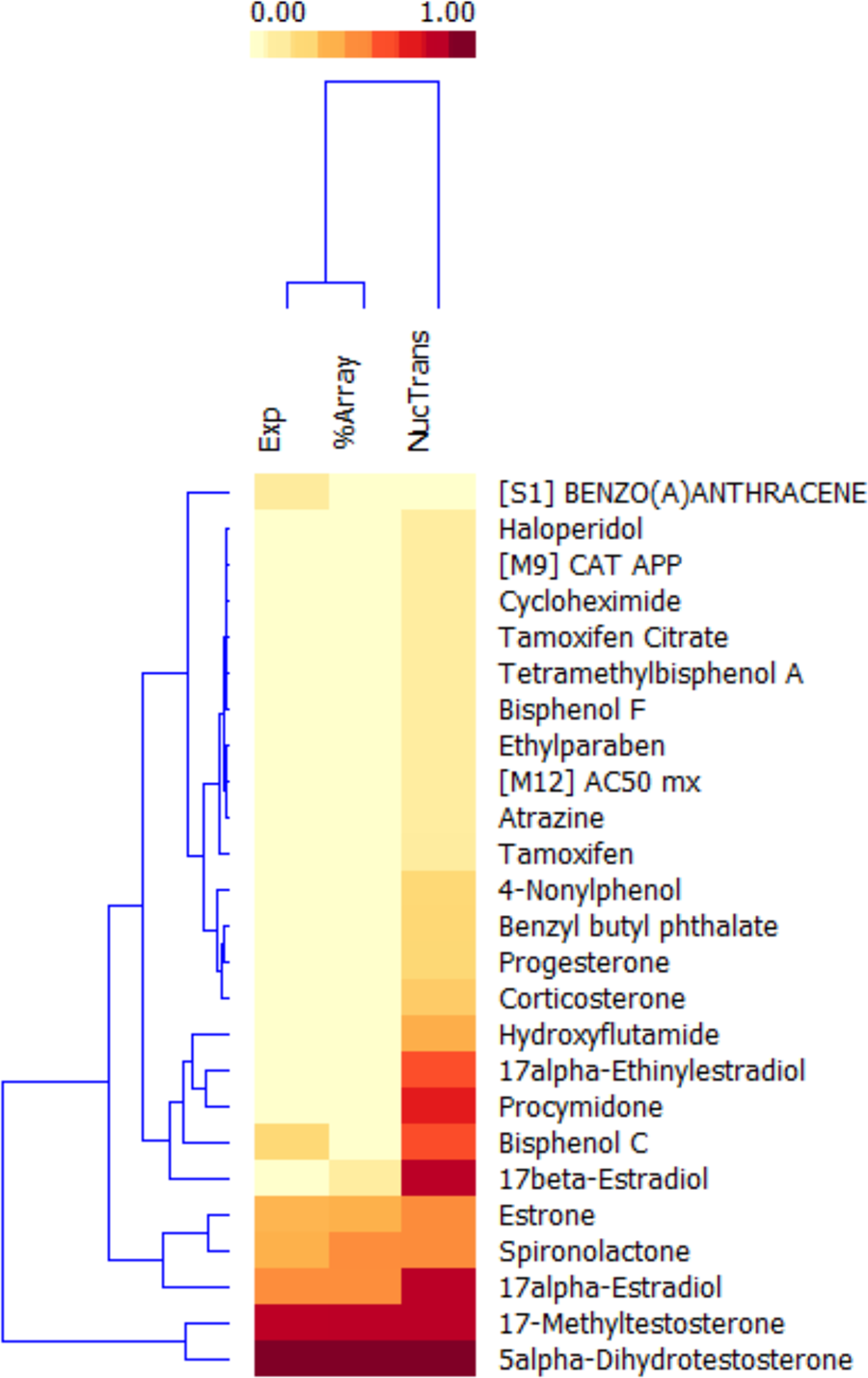
Unsupervised clustering of GFP-ARER endpoints of active chemicals. Normalized area under the curve (AUC) scores from active chemicals identified in either the Environmental Protection Agency or Agency for Toxic Substances and Disease Registry reference sets were clustered using a K-means–based algorithm. All inactive scores (<0.1) were set to zero to improve clustering performance and visualization. Both the chemicals and endpoints were clustered.
Cluster 2 contains three chemicals that induced a low/moderate AUC score for all three metrics. Spironolactone has demonstrated mixed in vitro and in vivo (anti-)androgenic activity in previous studies. 36,37 Estrone has previously demonstrated in vitro androgenic activity. 37 A literature review classified 17alpha-estradiol as inactive; however, an EPA computational model predicting androgenic activity based on multiple in vitro assays identified this chemical as a moderate/weak androgen. 36 This suggests cluster 2 compounds are those with mixed/weak androgenic activity in in vitro assays.
Cluster 3 features chemicals that predominantly have a moderate to weak nuclear translocation AUC score. Those chemicals with stronger AUC scores include known anti-androgens (bisphenol C, procymidone, hydroxyflutamide, 17alpha-ethynilestradiol).
36
-38
E2 (17beta-estradiol) also demonstrates a moderate AUC score despite being generally considered inactive with regard to (anti-)androgenic activity.
36,37
Chemicals with weak translocation AUC scores include several that have demonstrated anti-androgenic activity (progesterone, corticosterone, benzyl butyl phthalate) but largely consist of chemicals with unknown (anti-)androgenic potential. A summary of chemical information, previously reported activity, and cluster assignment can be found in
As a proof-of-concept demonstration of how the GFP-ARER: PRL-HeLa cell model may be used as an androgen biosensor, we screened a panel of 44 environmental samples collected at 24 unique sites of a major shipping channel/industrialized area in Texas as part of an EPA-funded Houston Shipping Channel/Galveston Bay Texas A&M University Superfund Research Program. Samples were prepared and concentrated from sediment samples using a cyclohexane-DMSO extraction method (see the Methods section) and tested at 1:200 to 1:200,000 dilutions. Cells were treated for 2 h, and activity was compared to measure the AUC response from DHT at concentrations ranging between 0.01 nM and 10 nM. K-means cluster analysis of results identified three samples with moderate/weak induction of GFP-ARER nuclear translocation, chromatin binding, and receptor stabilization ( Fig. 5A , B ). These samples demonstrated 9% to 33% of DHT activity. An additional 13 samples induced nuclear translocation by achieving only 10% to 29% of the DHT response. Active samples were dispersed throughout the sampled region ( Fig. 5C ); however, further analysis will need to be done using orthogonal approaches to verify activity. Importantly, the total processing time of samples (cell treatment, imaging, and data analysis) was less than 12 h, indicating that the cell model may be applied to rapidly measure potential EDC activity and identify areas of concern.
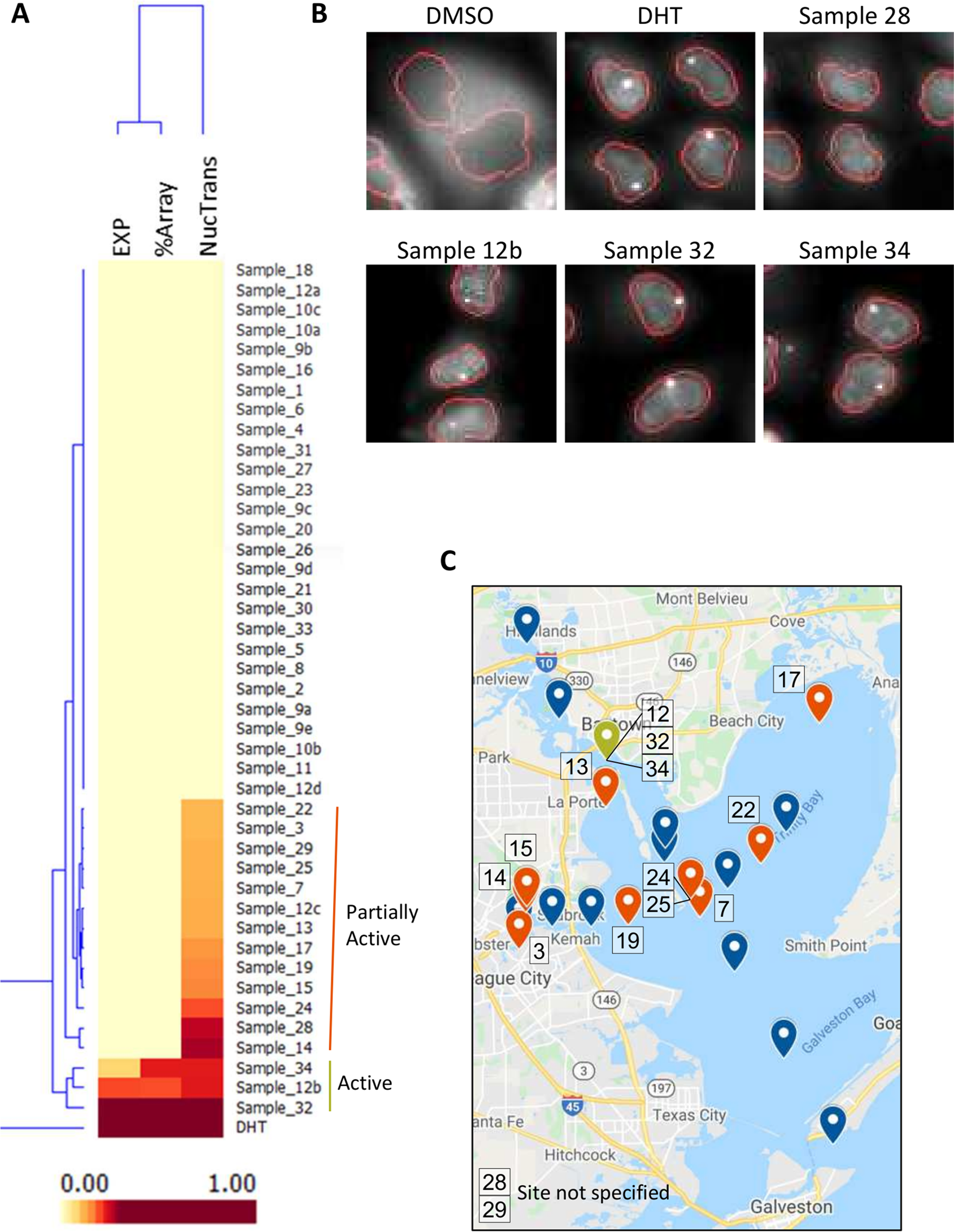
GFP-ARER used to detect potential (anti-)androgenic activity in sediment samples. GFP-ARER: PRL-HeLa cells were seeded onto 384-well plates and induced with 200 ng/mL doxycycline for 24 h prior to treatment. Cells were treated with 1:200 to 1:200,000 dilutions of cyclohexane-DMSO extracted samples collected from 24 sites around the Houston, Texas, shipping channel. Cells were then processed for fluorescence and imaged using the Molecular Devices ImageXpress at 20× magnification. (
Discussion
We have taken the established image-based high-throughput GFP-ER: PRL-HeLa assay for estrogenic activity and adapted it to detect (anti-)androgenic activity by exploiting the modular nature of NRs. The GFP-ARER: PRL-HeLa cell model allows the direct visualization of multiple mechanistic steps of AR genomic signaling at the single-cell level. By swapping the AR DBD for that of ER, the GFP-ARER–based model allowed for the single-cell quantification of (anti-)androgen–induced nuclear translocation, chromatin binding at an ERE-rich locus, coregulator recruitment, and transcriptional activity. We were also able to observe androgen-induced increases in total GFP signal per cell (AR stabilization), consistent with an established agonist-induced decrease in the degradation rate of AR. 39 Although a fraction of the total increase in GFP intensity may be due to consolidation of a diffuse cytoplasmic signal into the nuclear compartment as nuclear translocation occurs, we did not see similar increases in total GFP signal with the bicalutamide antagonist, which also induces nuclear translocation.
These are similar endpoints as used in the GFP-ER: PRL-HeLa model and recapitulate those measured using multiple biochemical and molecular methods, generally at the population level. Importantly, the observed responses occur rapidly, with nuclear translocation and chromatin binding occurring in less than 60 min and complete response to DHT observed within 120 min (
One key difference observed between the GFP-ARER cell model and the GFP-ER cell model is the ability of antagonist to induce visible receptor chromatin binding. In the GFP-ER model, selective estrogen modulators and pure ER antagonists such as fulvestrant (ICI 182 780) induce small visible arrays, indicative of chromatin binding with minimal chromatin remodeling. 13,14,18,19 In contrast, the AR antagonists bicalutamide and hydroxyflutamide induced GFP-ARER nuclear translocation but undetectable evidence of chromatin binding at the ERE-rich PRL locus. Antagonist-induced nuclear translocation of endogenous wild-type AR in prostate cancer cells is well established. 41 In addition, genome-wide ChIP-seq approaches have confirmed that both antagonist-bound ER and AR can bind some, but not all, chromatin-binding sites associated with agonist-induced transcriptional regulation. 42,43 This suggests that the ERE-rich PRL array resembles a promoter region recognized by both agonist- and antagonist-bound receptor in the GFP-ER cell model, whereas the PRL array resembles a region recognized thus far only by androgen-bound receptor in the GFP-ARER cell model. Interestingly, in the GFP-PRER: PRL-HeLa cell model, the PR antagonist RU486 induces visible chromatin binding whereas ZK98299 does not, suggesting that further screening may yet identify AR anti-androgens that result in stable chromatin binding by GFP-ARER at the PRL array. 28
The ligand-dependent array binding of AR highlights how the GFP-ARER: PRL-HeLa cell model may represent a beneficial compromise between rapid transcriptional reporter assays and slower androgen-dependent growth assays. Although reporter assays are fundamental for the high-throughput analysis of potential EDC activity, they measure AR activity within the context of a single promoter (either endogenous or synthetic) driving reporter gene expression. There is yet no defined minimal set of AR-responsive genes known to faithfully predict in vivo (anti-)androgenic EDC activity, and these assays are vulnerable to nonspecific results because of the coexpression of other NRs. 11 The GFP-ARER: PRL-HeLa cell model also relies on the single PRL promoter construct integrated into the PRL-HeLa cell line as a promoter array; however, the direct visualization of GFP-ARER chromatin binding (and transcriptional activity via mRNA FISH) minimizes the risk of nonspecific results. Further, the multiplex nature of the assay also allows the quantification of global effects on AR signaling such as nuclear translocation, subnuclear organization, and expression levels. This is similar to our earlier efforts using GFP-AR expression in HeLa cells, and others have advanced image-based AR bioassays to efficiently and inexpensively monitor androgenic activity from multiple types of samples, including urine samples, in the detection of anabolic steroid abuse. 32,40,44,45
Although the GFP-ARER: PRL-HeLa cell model has advantages over traditional methods for detecting androgenic activity, challenges remain. Reliable detection of chromatin binding by GFP-ARER is dependent on the quality of sample preparation, requires high-resolution automated imaging platforms, and, as with most high-content assays, is dependent on custom image analysis and data analysis algorithms. Although the pattern of nuclear translocation with negligible specific chromatin binding suggests anti-androgen–like activity, additional experiments are required to more directly detect the inhibition of DHT-induced responses. To avoid further complexities of downstream effects from ligand exposure and fit the assay within a high-throughput context, short-term experiments were the logical focus, which naturally precluded observations of responses requiring more time. Cell proliferation assays for the detection of estrogenic EDC activity are well established, simple, and sensitive. Because cell proliferation assays typically last for days/weeks, they potentially allow the detection of active metabolites or other important secondary changes that may not be detected in a high-throughput assay focusing on early (∼1–2 h) mechanistic steps in receptor activation. However, relatively few androgen-dependent cell proliferation assays have been developed and tend to rely on cell lines that express AR’s harboring point mutations known to alter ligand specificity. 45
Relatively little is known about the potential (anti-)androgenic activity of the many thousands of manmade chemicals in the environment. There is growing concern that a number pesticides, flame retardants, packaging, and industrial chemicals may harbor AR bioactivity that could potentially affect human health. 14,16,36,37 Through our participation in the Texas A&M Superfund Research Program, we were able to successfully detect androgenic activity in sediment samples collected from the Houston Ship Channel and Galveston Bay. The sediment samples provide a depth-dependent historical record of environmental contamination of this highly urbanized region containing markedly busy seaports and numerous permitted industrial sites located throughout the watershed. In this analysis, we did not detect androgenic activity in samples from the Galveston Bay, which included sampling sites farthest from the watershed. This suggests that higher concentrations of androgenic chemicals are rapidly dispersed with distance. Identifying the androgenic chemicals and their potential sources is an ongoing project within the Superfund Research Program. In addition, we are in the process of developing an anti-androgenic protocol using the GFP-ARER cell model to improve our ability to distinguish between weak androgens and anti-androgens.
It is clear that adoption of high-throughput in vitro screening assays have enabled the development of computational models capable of predicting the in vivo activity of endocrine disruptors. 36,46 A recent androgenic model relies on 11 different assays spanning three assay types (hormone binding, coactivator protein complementation, transcriptional reporter) to achieve 95.2% to 97.5% predictive accuracy. 36 The GFP-ARER: PRL-HeLa cell model rapidly measures three additional mechanistic steps of AR signaling (nuclear translocation, specific chromatin binding, receptor stabilization) in a single assay that would complement those already being used in computational models. Continued studies are warranted to further explore assay performance and classification models using additional EPA-validated reference chemicals and additional real-world samples (e.g., water samples from flooded areas). A meritorious aspect of expanding the use of the PRL-HeLa model with chimeric NRs is the fact that functional posttranslational modification sites are retained, thus allowing further exploration of activation/inhibition of cell-signaling pathways that could alter AR signaling in cancer research. In conclusion, we have developed the GFP-ARER: PRL-HeLa cell model that allows for the rapid quantification of multiple mechanistic aspects of AR signaling. This adds to our library of PRL-HeLa cell models based on ERα, ERβ, and PRER expression, expanding the versatility of the assay platform and enhancing the ability to identify molecules and cell-signaling pathways that alter NR function and potentially protecting and improving human health.
Supplemental Material
Supplemental Material, Szafran_Mancini_ARER_Supplemental_2_0 - A Mechanistic High-Content Analysis Assay Using a Chimeric Androgen Receptor That Rapidly Characterizes Androgenic Chemicals
Supplemental Material, Szafran_Mancini_ARER_Supplemental_2_0 for A Mechanistic High-Content Analysis Assay Using a Chimeric Androgen Receptor That Rapidly Characterizes Androgenic Chemicals by Adam T. Szafran, Michael J. Bolt, Caroline E. Obkirchner, Maureen G. Mancini, Christine Helsen, Frank Claessens, Fabio Stossi and Michael A. Mancini in SLAS Discovery
Footnotes
Acknowledgments
We thank for Dr. Moczygemba and the Flow Cytometry and Cell Sorting Facility (FCCSF) located at the Texas A&M University Institute of Bioscience and Technology. The authors are also indebted to Keith Houck, PhD (EPA), for coordinating access to reference chemicals used in this study. Finally, we would like to thank members of the Texas A&M Superfund Research Program, especially the Project 1 research group of Dr. Knap that collected the sediment samples, and Zunwei Chen from Dr. Rusyn’s lab in Project 3 for performing the sediment extraction and delivering reference chemicals and samples to Baylor College of Medicine.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: M.A.M., F.S., and A.T.S. are funded by NIEHS (P42ES027704, principal investigator [PI], Rusyn). M.A.M and F.S. are supported via the CPRIT-funded GCC Center for Advanced Microscopy and Image Informatics (RP170719; PI, Mancini). Imaging is also supported by the Integrated Microscopy Core at Baylor College of Medicine with funding from the National Institutes of Health (DK56338 and CA125123), CPRIT (RP150578), the Dan L. Duncan Comprehensive Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics.
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.