
Abstract
The targeted delivery of potent cytotoxic molecules into cancer cells is considered a promising anticancer strategy. The design of clinically effective antibody–drug conjugates (ADCs), in which biologically active drugs are coupled through chemical linkers to monoclonal antibodies, has presented challenges for pharmaceutical researchers. After 30 years of intensive research and development activities, only seven ADCs have been approved for clinical use; two have received fast-track designation and two breakthrough therapy designation from the Food and Drug Administration. There is continued interest in the field, as documented by the growing number of candidates in clinical development. This review aims to summarize the most recent innovations that have been applied to the design of ADCs undergoing early- and late-stage clinical trials. Discovery and rational optimization of new payloads, chemical linkers, and antibody formats have improved the therapeutic index of next-generation ADCs, ultimately resulting in improved clinical benefit for the patients.
Introduction
Approximately a hundred years have passed since Paul Ehrlich proposed the concept of antibodies as the “magic bullet” for selective targeting of malignant cells. 1 More recently, progress in hybridoma technologies and recombinant DNA techniques has enabled the generation of highly effective monoclonal antibodies (mAbs) 2–8 to be developed for therapeutic purposes. However, in spite of the conceptual simplicity of antibody–drug conjugates (ADCs) as a therapeutic modality, the design, generation, and clinical use of these therapies have proved challenging. After 30 years of intense research and development activities, only seven ADCs have been granted clinical approval ( Table 1 ), while about 70 ADCs are undergoing clinical trials ( Table 2 ).
Marketed ADCs.
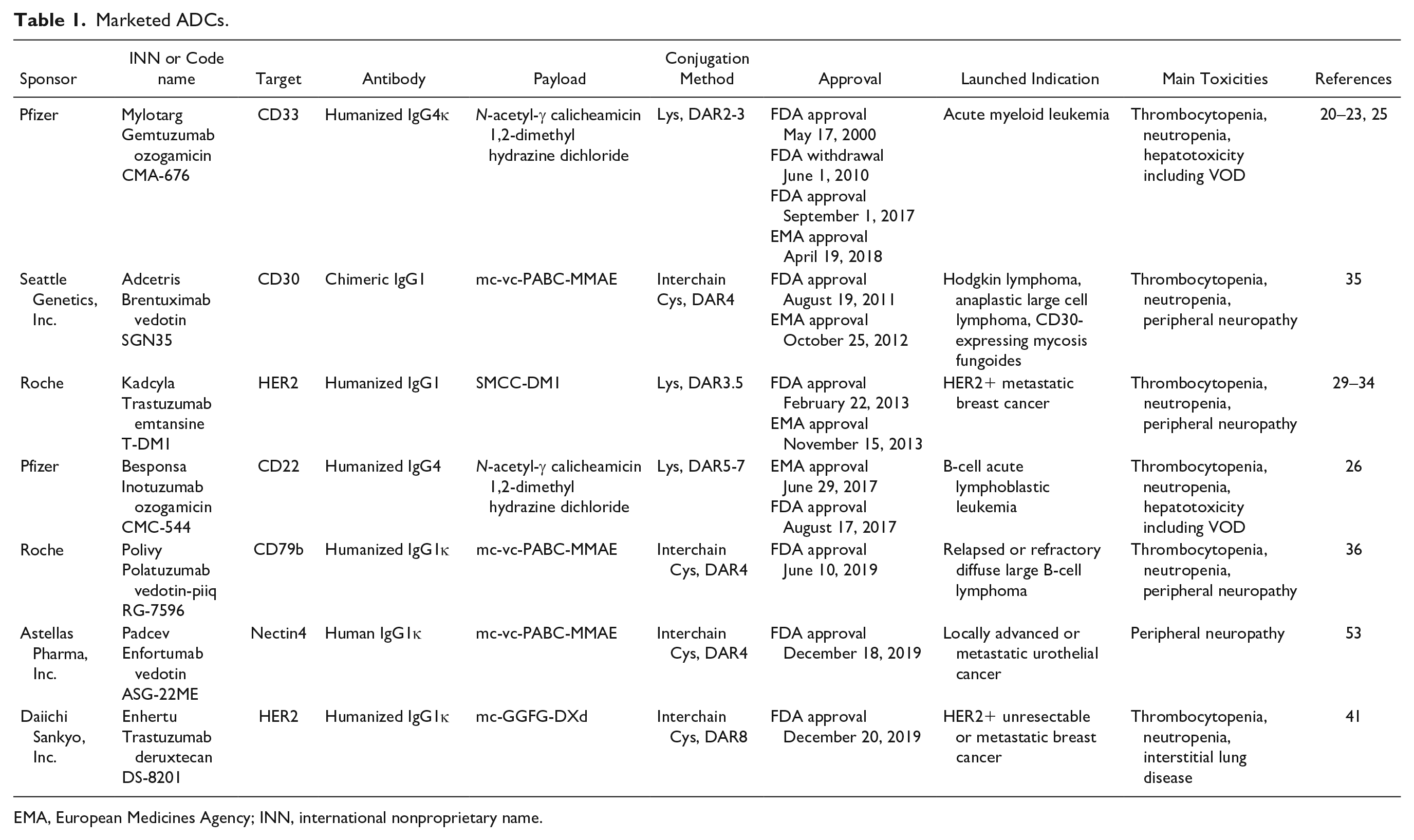
EMA, European Medicines Agency; INN, international nonproprietary name.
Clinical Stage ADCs.
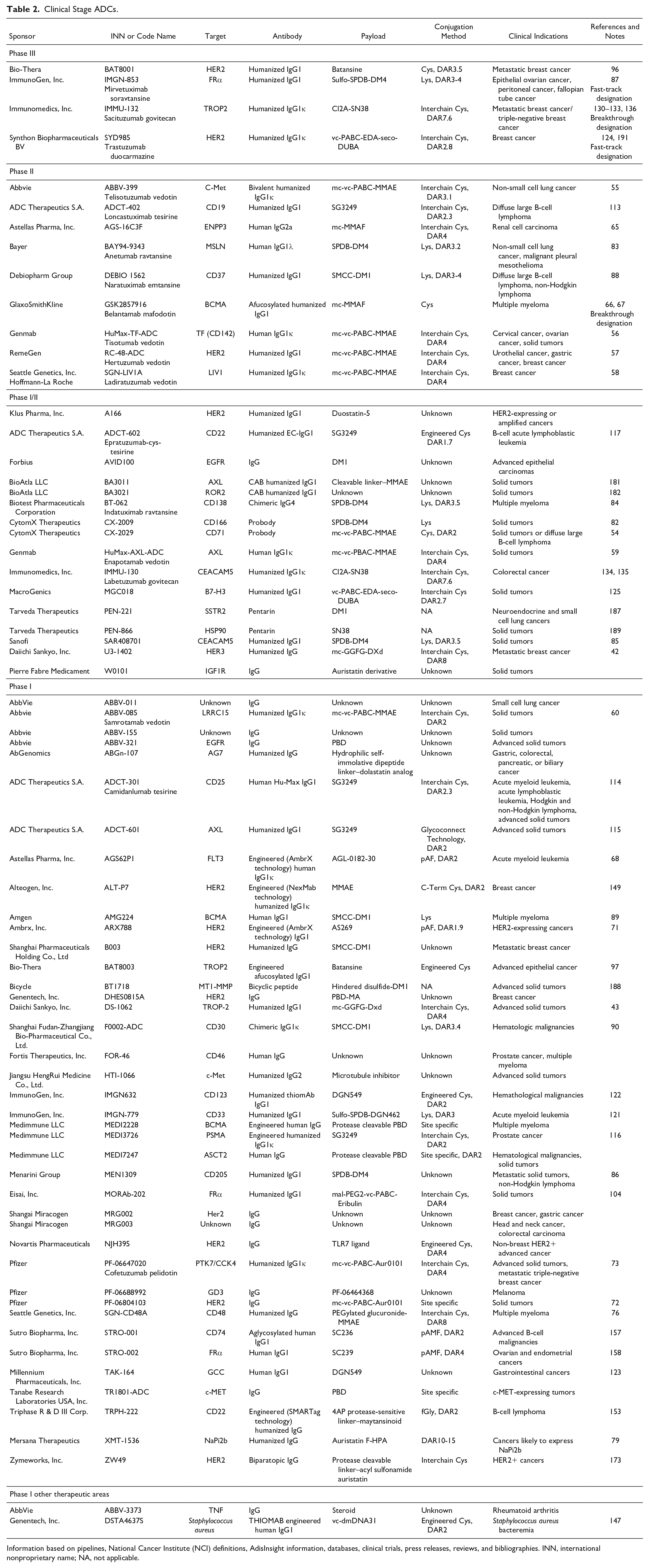
Information based on pipelines, National Cancer Institute (NCI) definitions, AdisInsight information, databases, clinical trials, press releases, reviews, and bibliographies. INN, international nonproprietary name; NA, not applicable.
The initial appeal of ADCs as a concept was the theoretical improvement they afforded to antibody therapies, in addition to increasing the therapeutic index of highly potent cytotoxic compounds.9–12 Indeed, the conjugation of small molecules to tumor-selective antibodies is intended to increase their specific retention and cytotoxic activity at the site of tumors, while avoiding activity on healthy tissues. The combination of these actions is to limit undesirable side effects commonly seen with conventional chemotherapeutic drugs.
ADCs are prodrugs (
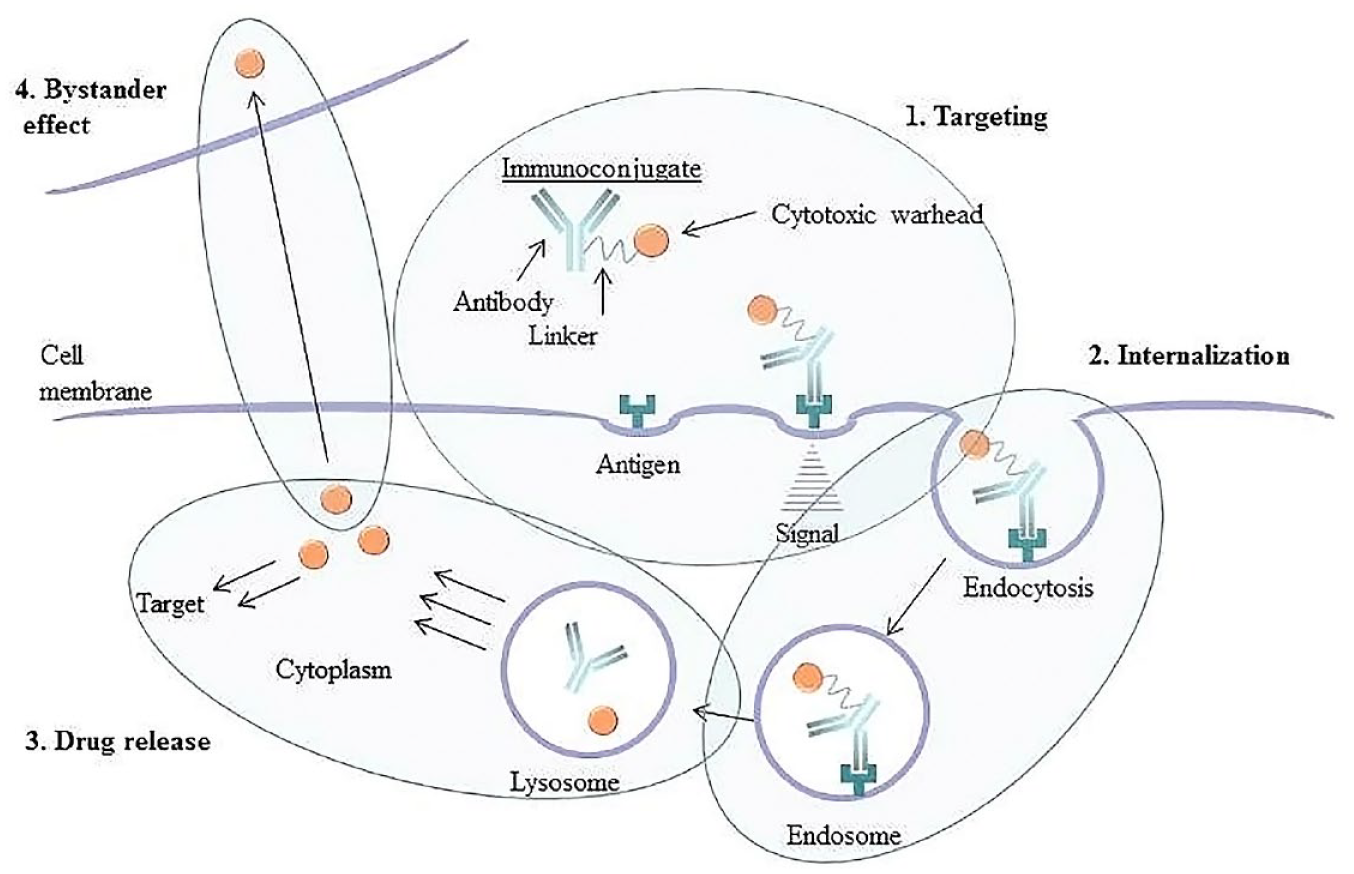
Mechanism of action of an ADC.
Today, ADCs have yet to live up to their expected clinical potential, and it is anticipated that a better understanding of the mechanisms of ADC toxicity and efficacy will help in the design of better drugs and improve clinical design, resulting in reduced clinical attrition rates. Key parameters to consider for ADC optimization include:
The presence of a tumor-specific antigen, homogeneously and highly expressed at the tumor site, resulting in efficient internalization and intracellular trafficking of the ADC upon antibody binding. Lower levels of expression may be compensated by increased internalization and trafficking efficiency.
The drug carrier (the antibody) with optimal tumor selectivity and retention properties, as well as sufficient persistence in the blood. As described previously, another key parameter that defines ADC efficacy is the efficiency of drug release. This parameter is dependent on internalization and the trafficking efficiency of the antibody–antigen pair to the appropriate subcellular compartments.
The linker and cytotoxic agent define the properties of the metabolite that is intended to act against the specific cancer type. The metabolite derived from the ADC should have potent cell-killing properties. The indication should also be intrinsically sensitive to the cytotoxic metabolite of the ADC.
The conjugation technology, such as SSC, optimized to improve efficacy, maximize pharmacokinetic (PK) exposure, and limit toxicity and payload metabolism in circulation while attached to the antibody. 17
An adapted clinical trial design, focusing on the dosing schedule and the drug combinations in order to improve the therapeutic index of the agent.
After a brief overview of the ADC design of the Food and Drug Administration (FDA)-approved molecules, this review primarily focuses on innovations that feature in ADCs currently in clinical trials. These innovations comprise the development of new payloads, chemical linkers, and antibody formats that have been used to maximize the therapeutic index of the next-generation ADCs currently showing increased clinical benefit for patients. The success of the targeted drug delivery approach also relies on the identification and validation of appropriate antigenic targets, a subject that is not covered in this article, as several excellent reviews already exist on the topic.10,13,18,19
FDA-Approved ADCs
To date, seven ADCs have received marketing authorization ( Table 1 ). Four of these ADCs have been approved for hematological malignancies, where the target tends to be more readily accessible, with homogeneous expression of surface antigen, compared with those of solid tumors. The remaining ADCs are approved for the treatment of solid tumors, where poor tumor tissue penetration and heterogeneous antigen distribution are additional challenges for the successful clinical efficacy of ADCs targeting solid tumors.
Mylotarg (gemtuzumab ozogamicin; Pfizer, New York City, NY) was the first ADC to reach the market through accelerated FDA approval in 2000, after it was fast-tracked for the treatment of acute myeloid leukemia (AML). 20 This molecule is an anti-CD33 IgG4 antibody covalently linked to the cytotoxic agent N-acetyl γ-calicheamicin, via a pH-sensitive hydrazone linker. N-acetyl γ-calicheamicin is a DNA-damaging agent from the enedyine family. This class of compound induces DNA double-strand breaks and subsequent cancer cell death. 21 The key insight obtained from the use of this agent is that clinical efficacy can be achieved, with limited side effects, by using a highly cytotoxic compound (100–1000 times more cytotoxic than usual chemotherapeutic agents) coupled with a tumor-selective humanized antibody. In spite of providing these insights, Mylotarg was withdrawn from the market 10 years after its initial release over safety concerns raised during a confirmatory trial. 22 This study (SWOG S0106) highlighted increased incidence of mortality during the induction phase, with severe hepatic toxicity, as well as increased risk of potentially fatal veno-occlusive disease (VOD) observed in study patients, particularly those who had undergone hematopoietic stem cell transplant. Ultimately, no clinical benefit was observed for this agent. 23 The limited therapeutic index was the result of the observed toxicities, attributed to inappropriate circulatory release of the free drug, 24 due to the instability of the pH-sensitive hydrazone linker in patient blood. This hypothesis has been challenged. In a recent phase III study, clinical benefit and an improved toxicity profile were observed for a subpopulation of acute myeloid lymphoma patients, following fractionated dosing. These observations suggested that toxicity was being driven by a Cmax effect and led to the new approval of this ADC for a restricted CD33-positive AML patient population in 2017. 25
Another ADC, Besponsa (inotuzumab ozogamicin; Pfizer), with the same calicheamicin payload, was also approved in 2017. This ADC is composed of a humanized IgG4 mAb against CD22, which is covalently linked to the same cytotoxic agent N-acetyl γ-calicheamicin via an acid-cleavable linker, and stochastically conjugated to the lysines of the antibody. The FDA granted this ADC’s approval for use as a single agent in adults with relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia (ALL), the most common type of adult ALL. 26 Similar adverse events occurred during the clinical trials of both ADCs and suggested that these effects were not target mediated but linked to the calicheamicin-linker platform. Besponsa combination therapies are currently being evaluated in the phase I/II or II setting in ALL and chronic myeloid leukemia and in the phase I setting in Burkitt’s lymphoma.
Adcetris (brentuximab vedotin; Seattle Genetics, Inc., Bothell, WA, and Takeda, Osaka, Japan), Kadcyla (trastuzumab emtansine; Genentech, Inc., South San Fransisco, CA), Polivy (polatuzumab vedotin-piiq; Genentech, Inc.), and Padcev (enfortumab vedotin-ejfv; Astellas Pharma US, Inc., Northbrook, IL) were launched in 2011, 2013, and 2019 (for the last two), respectively. All four ADCs are based on the tubulin binders MMAE (auristatin) and DM1 (maytansinoid). Auristatins are synthetic analogs of dolastatin 10, which inhibits tubulin polymerization, causing cell cycle arrest and eventual death. This agent is 10–50 times more potent than other tubulin-interacting agents such as vinblastine. 27 Maytansinoids are maytansine derivatives and share the same destabilizing microtubule assembly 28 mechanism of action as the vinca-alkaloids. Kadcyla is composed of a humanized HER2-directed mAb trastuzumab (Herceptin; Genentech, Inc.) randomly conjugated through lysines to maytansinoid cytotoxic DM1 molecules, via a noncleavable thioether linker (N-succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate; SMCC), with an average DAR of 3.5. 29 Kadcyla is approved as a single agent for the treatment of patients with HER2-positive metastatic breast cancer who have previously received Herceptin and taxane chemotherapy, separately or in combination,30,31 and for the adjuvant treatment of patients with HER2-positive early breast cancer who have residual invasive disease after neoadjuvant taxane and trastuzumab-based treatment. 32 This is the first ADC that has been approved in a solid tumor indication. The superior efficacy of Kadcyla was demonstrated for patients presenting residual invasive HER2-positive breast cancer at surgery, after neoadjuvant therapy containing a taxane and trastuzumab, but not in the neoadjuvant first-line setting. 33 The success of Kadcyla is likely due to its mechanism of action, consisting of the combined antitumor effects of trastuzumab (antibody-dependent cytotoxic activity [ADCC]-mediated cytotoxic effect) and cytotoxic Lys-SMCC-DM1 metabolite released within the target cells upon ADC processing in the lysosome. The reasons for the failure as a front-line agent in neoadjuvant therapy remain unclear. 34
Adcetris is a CD30-directed chimeric mAb coupled via a cleavable traceless linker, including a para-amino benzyl carbamate (PABC) self-immolative spacer, to the auristatin derivative MMAE. Adcetris received accelerated approval for the treatment of relapsed or refractory CD30-positive Hodgkin lymphoma (HL) as well as for patients with systemic anaplastic large cell lymphoma (ALCL), after prior failure of at least one multiagent chemotherapy regimen. 35 Combinatorial studies of Adcetris are expected to enlarge its use in the treatment of classical Hodgkin lymphoma or other CD30-positive hematological malignancies.
Polivy is a CD79b-directed ADC used in combination with bendamustine and rituximab 36 for adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), after at least two prior therapies.
Padcev is a Nectin4-directed ADC used in the treatment of adult patients with locally advanced or metastatic urothelial cancer, who have previously received a programmed cell death receptor-1 (PD-1) or programmed cell death ligand 1 (PD-L1) inhibitor antibody and a platinum-containing chemotherapy. It reached the market through accelerated FDA approval in 2019, after having received breakthrough designation for the treatment of metastatic bladder cancer.
Adcetris, Polivy, and Padcev share the same auristatin-derived payload, namely, mc-vc-PABC-MMAE, whose maleimidocaproyl (mc) cleavable linker is cathepsin B sensitive. The three ADCs are conjugated to interchain cysteines with an average DAR of 4. After internalization of the ADC into target cells, the released drug can elicit bystander effect because of its inherent physicochemical properties.
Enhertu is the second ADC, with Kadcyla, composed of HER2-directed mAb trastuzumab (Herceptin; Genentech, Inc.). The major difference between Kadcyla and Enhertu is the payload consisting of an agent with topoisomerase I inhibitor activity, mc-GGFG-Dxd, 37 based on exatecan (or Dxd).38–43 Dxd is a derivative of DX-8951, a novel topoisomerase I inhibitor that has more potent efficacy than irinotecan against various tumor xenograft models, including irinotecan-resistant tumors, and it was developed up to phase III.44,45 This payload encompasses a stable protease-sensitive cleavable linker that releases Dxd, having a primary hydroxyl group. 39 Moreover, Enhertu is a DAR8 ADC, which is different from other approved ADCs. It is used in the treatment of patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting. It is the latest ADC to reach the market through accelerated FDA approval in 2019, after having received breakthrough designation for the treatment of breast cancer.
Clinical trials with several ADCs bearing the same tubulin binder payload and linker have led to the hypothesis that the dose-limiting toxicities are predominantly unrelated to the target antigen, but rather driven by the drug/metabolites of the ADC. 46 There is a clear association between tubulin binder payloads and the development of reversible ocular side effects, although the frequency and severity of these events are related to the chemical nature of the formed metabolites. For example, MMAE-containing ADCs show less frequent ocular toxicities than MMAF-containing ADCs, a noncleavable tubulin binder ADC derived from auristatin. By contrast, the toxicities of most MMAE conjugates in clinical trials or on the market share a similar toxicity profile characterized by thrombocytopenia, neutropenia, and peripheral neuropathy. 47
Most of the current ADCs that have reached the clinic still possess a relatively narrow therapeutic window, because of off-target toxicities, competition with unconjugated antibodies, and aggregation or fast clearance of high DAR ADCs. For example, for Adcetris and Kadcyla, there are approximately 5% unconjugated species that compete with the drug-loaded species for binding to the antigen. For Mylotarg, the instability of the linker allows for up to 50% of unconjugated species in circulation. 48 In specific cases, it has also been demonstrated that species with higher DAR could lead to lower tolerability and higher plasma clearance.19,49,50
It is becoming clearer that, at least in the case of ADCs with payloads acting primarily on dividing cells, such as tubulin binders, the therapeutic index is primarily influenced by the nature of the payload–linker and the physicochemical properties of its metabolites more so than the expression characteristics of the antigen on the tumor surface versus normal tissues. In addition, stochastic conjugation, the level of unconjugated antibody species in the circulation, and limitations of antibody formats with respect to their biodistribution in solid tumors are other parameters affecting the clinical efficacy of ADCs. In parallel, clinical scientists are working on translational strategies such as the optimization of dosing regimen, the use of biomarkers for the selection of patients who may best benefit from treatment, the early visualization of response signals, and potential synergistic combination therapies (for a review, see Coats et al. 51 ).
Innovation under Conventional ADC Format
Development of New Cytotoxic Payloads
The first ADCs derived from coupling conventional chemotherapy agents to antibodies (e.g., doxorubicin immunoconjugate BMS-182248-1
52
) displayed disappointing results in the clinics and were deemed to be insufficiently potent for this approach. As a consequence, much more potent agents (calicheamicin, maytansine, and auristatin) became the payloads of choice. To overcome limitations of the first generation of ADCs based on these agents, two approaches have been deployed to increase payload diversity: First, payloads sharing the mechanisms of action of the two clinically validated warheads, namely, DNA-damaging agents and microtubule-binding agents, were optimized in order to further improve the ADC therapeutic index. Second, payloads with alternative mechanisms of actions were explored, for example, topoisomerase I inhibition (
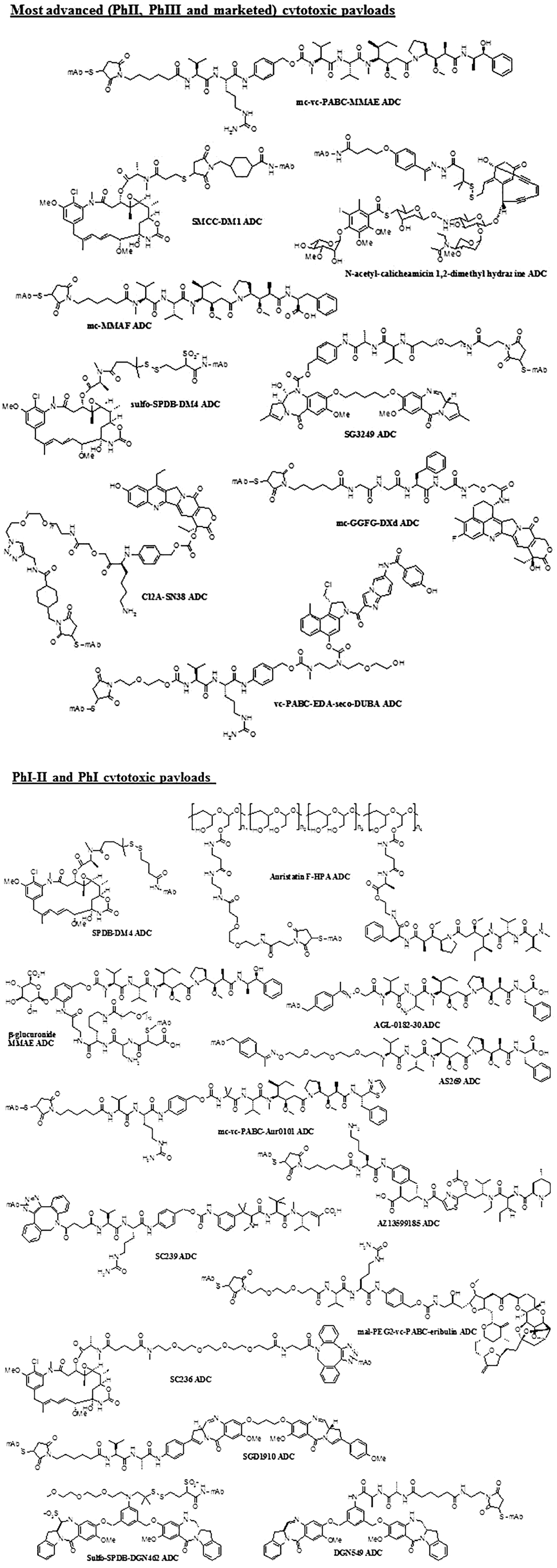
Cytotoxic payloads for ADCs at the clinical stage.
For the auristatin series of tubulin binders, Seattle Genetics developed mc-MMAF, a noncleavable payload based on the auristatin MMAF, as an alternative payload to the first-generation cleavable mc-vc-PABC-MMAE.53–60 MMAF is less cell permeable than MMAE, and thus less cytotoxic as an unconjugated drug. However, this agent turned out to be highly active as an ADC. 61 In spite of this finding, ABT-414,62–64 an EGFR-directed ADC based on the mc-MMAF payload, recently failed to meet the primary endpoint of overall survival in phase III trials, and as a result, enrollment in all current studies has been halted. Nevertheless, the payload is still under clinical evaluation in earlier phases, as the payload for GSK2857916 and AGS-16C3F.65–67 Several other auristatin payloads, currently under clinical evaluation, with DAR values ranging from 2 to 15, have been developed to overcome some limitations of the cathepsine B-sensitive mc-vc-PABC-MMAE payload. These agents are:
AGL-0182-30 consisting of a noncleavable linker and an azido-valine-MMAF that has been modified to contain a N-[2-(aminooxy)acetyl] moiety at the N-terminus, to enable SSC to the antibody at a p-acetyl-phenylalanine (pAF) via an oxime. 68
AS269 amberstatin consisting of a noncleavable PEG linker designed to improve payload solubility and polarity, and to reduce the export of metabolites by efflux pumps. In addition, the presence of an alkoxyamine-containing MMAF group enables SSC to be carried out via a similar oxime.69–71
A payload consisting of a cleavable vc-PABC linker and Aur0101, a new microtubule inhibitor selected following N-terminal optimization of dolastatin 10.72,73 Introduction of α,α-disubstituted amino acids led to auristatin analogs retaining high in vitro potency, while exhibiting differential ADME properties, such as increased liver intrinsic clearance, compared with other auristatin payloads for ADCs. This feature is expected to improve the toxicity profile of the ADC. 74
A cleavable MMAE payload consisting of a hydrophilic PEGylated glucuronide linker75,76 that is cleaved by lysosomal β-glucuronidase to achieve efficient drug release. 77 This improvement of the payload physicochemical properties led to an ADC with a DAR value of 8, which exhibited increased potency without accelerated clearance in plasma compared with the DAR4 ADC. Furthermore, the maleimidocaproyl (mc) spacer, used for mAb conjugation and known to be prone to partial deconjugation, was also replaced by an optimized self-stabilizing maleimide (mDPR) that prevents circulatory payload deconjugation via rapid postconjugation thiosuccinimide hydrolysis. 78
Dolaflexin (or AF-HPA), 79 another auristatin payload, consists of several molecules of NMe-MMAF80,81 covalently linked to alcohol moieties of a water-soluble polyacetal-based polymer. The high level of water solubility of the polymer compensates for the hydrophobicity of the cytotoxic drug and enables DAR values of 12–15 to be reached, while maintaining acceptable plasma clearance.
The second group of tubulin binders are those belonging to the maytansine series. ImmunoGen Inc. (Waltham, MA) also developed disulfide cleavable payloads, SPDB-DM482–86 and sulfo-SPDB-DM4, 87 which, unlike SMCC-DM1,88–90 elicit bystander killing. 91 The optimal linker turned out to be target dependent for both cleavable and noncleavable classes of linkers.87,92,93 For disulfide cleavable linkers, varying the steric hindrance at carbon atoms adjacent to the disulfide linkage was one key parameter for balancing conjugate plasma half-life and efficient intracellular release of the cytotoxic drug, resulting in optimal in vivo activity. 94 The sulfo-SPDB-DM4 payload was explored in addition to more hydrophilic payloads in order to investigate the ability to produce maytansine ADCs with higher DAR values, compared with hydrophobic SPDB and SMCC linkers. As a result of its more polar nature, the active metabolite derived from the sulfo-SPDB-DM4 payload was anticipated to be a poor substrate for MDR efflux pumps, therefore overcoming the multidrug resistance of previous ADCs. 95 Batansine is another maytansinoid payload constituent in two new ADCs, BAT8001 96 and BAT8003. 97 Both of these ADCs are in clinical development and their structures have not been disclosed.
Besides the auristatin and maytansin payloads, other natural product-derived tubulin binder payloads have been developed for ADCs. Many groups have been working on tubulysin given its potent cytotoxicity against a wide range of human cancer cell lines, including significant activity in MDR cancer cell lines.17,98–100 The main challenge for this tubulysin series was to manage in vivo instability of the acetate, 101 a key structural element needed to attain significant potency. The tubulysin AZ13599185 progressed through phase I clinical trials as the payload for MEDI-4276, 98 a biparatopic HER2-directed ADC, but it was recently discontinued. The reason for discontinuation was a lack of observed benefit as a result of insufficient human exposure, due to the toxicity profile of the ADC’s warhead. 102
Various linker types and mAb conjugation chemistries were also explored when developing a new ADC payload from eribulin mesylate, marketed as Halaven. This drug is approved in the United States for the treatment of metastatic breast cancer. 103 The linker investigations led to the identification of maleimido-PEG2-vc-PABC-eribulin payload for MORAb-202. 104
Finally, the cryptophycin series, which is one of the most potent tubulin binder families ever discovered, has also been explored as a tubulin binder payload for ADCs.105,106 The payload is still in the preclinical stage of discovery. The main challenge with C52 ADCs was to address the deactivating species-dependent payload metabolism that was found to occur in mice. Switching from stochastic to SSC and optimizing the conjugation site reduced this mouse-dependent metabolism. 17 The deactivation was completely abrogated by designing new stable cryptophycin macrocycles, based on its metabolism pathway. 107
DNA-damaging agents, which differ from microtubule-binding agents in their ability to kill cells independently of the cell cycle stage, have also been extensively studied as payloads for ADCs. In addition to calicheamicin, dimers of pyrrolo-benzodiazepines (PBDs) and duocarmycins are the two chemical series that are the most exploited in this category of payload for ADCs. Dimers of PBDs are highly potent DNA minor groove cross-linkers and have been optimized using three different sites for linker introduction: the imine of one PBD monomer, the reactive moiety involved in DNA cross-linking; the exocyclic position of one PBD monomer; and the spacer in between the two PBD monomers. The most advanced representative of this family is Tesirine (SG3249). This agent is derived from the optimization of Talirine (SGD1910),108,109 which previously failed in clinical trials. The optimization strategy of Talirine aimed at lower payload hydrophobicity, while maintaining cytotoxicity. Optimized Tesirine110–117 consists of a less hydrophobic PBD dimer, SG3199, in which C2 aryl groups of SGD-1882 have been replaced by methyl groups and the tether between the two PBD monomers is lengthened by two methylene groups to offset the potency drop. The cathepsin B-cleavable valine–alanine–PABC linker was introduced on the imine of one PBD monomer instead of a C2 aryl group to mask the reactivity of the imine. Finally, payload solubility was further enhanced using a PEG8 spacer instead of the caproyl spacer. Tesirine is the payload for Rova-T111,112 and has reached phase III trials for small cell lung cancer. Rova-T showed no survival benefit compared with placebo in the MERU study, while in the TOHOE study this ADC demonstrated shorter overall survival compared with topotecan. As a result of these findings, Rova-T has been discontinued in clinical trials. However, Tesirine continues to be explored as a payload for other antibodies113–117 in clinical studies.
Another PBD dimer series still in preclinical studies consists of tomaymycin dimers, tomaymycin being a natural PBD. When using this agent, the linker site for conjugation is introduced on the spacer in between the two PBD monomers, 118 providing a symmetrical payload for the ADC. Finally, indolinobenzodiazepine dimers (IGNs), derived from PBDs, display three differentiating structural features:119,120 (1) the pyrrolidine ring is replaced by an indolino moiety to increase DNA binding and thereby potency, (2) the replacement of the alkyl tether by a phenyl spacer further improves potency and also provides a site for linker introduction, and (3) one imine is selectively reduced, resulting in an asymmetric monoimine DNA-alkylating dimer, in order to reduce payload toxicity, assumed to be related to the DNA-cross-linking mechanism of action. There are two payloads based on this monofunctional DNA-alkylating IGN: (1) DGN462 with a cleavable hydrophilic disulfide sulfo-SPDB linker and, to further improve payload solubility, a branched PEG3 moiety and a sulfonate group on the remaining imine;119,121 and (2) DGN549 with a cleavable alanine–alanine linker and a maleimido reactive group, enabling SSC to an engineered cysteine instead of stochastic conjugation on lysines.120,122,123
There are also ADCs containing a DNA-damaging payload belonging to the duocarmycin family124,125 in clinical trials. Duocarmycins are very potent DNA minor groove alkylators consisting of one DNA-binding and one DNA-alkylating moiety. The main challenges for developing this series as a payload for ADCs are its poor solubility and the stability of both the halogen-containing seco-prodrug used to reduce toxicity and the active cyclopropyl-containing spiro-form.124,126–128 Following extensive structure–activity relationship studies aimed at optimizing mainly the hydrophobicity of the core ring structure and the spontaneous elimination of the payload once released from the ADC, an imidazo[1,2-a] pyridine series was selected as the DNA-binding moiety of seco-DUBA (seco-DUocarmycin-hydroxyBenzamide-Azaindole). Linker introduction on seco-DUBA DNA-alkylating moiety was favored over introduction on the DNA-binding moiety as the simplest and most effective way to prevent premature ring closure to the spiro-form and inactivation of the conjugated drug in circulation. Finally, the linker of this optimized seco-DUBA payload consists of the cleavable valine-citrulline dipeptide, two self-immolative spacers—the PABC spacer and an additional bisamine cyclization spacer to allow for a carbamate linkage to seco-DUBA, as this type of linkage is usually more stable than a carbonate equivalent—and PEG2 moieties to further enhance payload solubility.
Topoisomerase I inhibitor-based payloads from the camptothecin series are the only compounds that display a mechanism of action distinct from tubulin and DNA-binding payloads. An alternative payload of this series to mc-GGFG-Dxd41–43 is Cl2A-SN38. The Cl2A-SN38 payload is derived from SN-38, the active metabolite of irinotecan, a clinically used chemotherapy drug.129–136 ADCs with the SN-38 payload differ from currently approved ADCs in three main aspects: (1) SN-38 is a moderately cytotoxic drug with nanomolar IC50 compared with the picomolar IC50 of other more potent agents; (2) SN-38 drug loading to the antibody is in the range of 6–8 versus lower substitution ratios for more potent cytotoxics (two to four drugs per antibody); and (3) the pH-sensitive carbonate linkage between the drug and the linker is moderately stable with a half-life in serum of ~24 h, enabling SN-38 to be released slowly at the tumor site once localized at target cells. This phenomenon makes the drug accessible not only to tumor cells internalizing the ADC, but also to surrounding cells. The linker is also composed of a PEG8 moiety to ensure sufficient payload solubility.
Other mechanisms of action for ADC payloads are currently being evaluated at the preclinical stage. 137 They include RNA splicing inhibition using natural product-derived thailanstatins 138 and RNA polymerase I inhibition using α-amanitin derivatives. 139 Both mechanisms are effective on both dividing and quiescent cells. Of note, α-amanitin is a hydrophilic natural product exhibiting potent cytotoxic activity in the ADC format, while, unconjugated, it is unable to enter normal cells via passive uptake. Another interesting feature of this series lies in the fact that hemizygous deletion of p53/Pol2RA increases the susceptibility to α-amanitin ADC and could thus be used as a potential biomarker.
Innovation Related to Antibody Modification
A number of key antibody features have been identified as playing an important role and are currently used in the design of clinical-stage ADCs. A high degree of specificity of the antibody for the tumor antigen is essential. An antibody that cross-reacts with other antigens or displays general nonspecific binding can be taken up into normal tissues in significant amounts, resulting in both toxicity and removal/elimination of the ADC before it can reach the tumor. 140 The antibody must also bind the target antigen with reasonable affinity (Kd < 10 nM) for efficient uptake into target cells, and it should be minimally immunogenic. It is also important to select an antibody with optimal PK properties (longer half-life with slower clearance in plasma 141 ) and desired immune-effector functions. These latter properties can be tailored through the use of different IgG isotypes and mutations in the Fc portion of the antibody.142,143 More recently, innovations related to the site of conjugation of the payload on the antibody have been addressed.
Site-Specific Conjugation
All the approved ADCs currently on the market exploit stochastic conjugation on preexisting amino acids with chemically activatable side chains, namely, lysine and cysteine. Lysine side chains have been used for the conjugation of calicheamicin and DM1 payloads, whereas those of cysteine have been used for the conjugation of the MMAE payload. More generally, the maytansin platform, including DM4, was developed through lysine conjugation chemistry, while auristatin payloads, including MMAF, were developed through cysteine conjugation. While stochastic conjugation methods have been used for most phase II and III compounds, more recent ADCs entering phase I are primarily conjugated using site-specific methods ( Table 2 ).
Regardless of the conjugated amino acid (cysteine or lysine), ADCs resulting from stochastic conjugation are heterogeneous products (
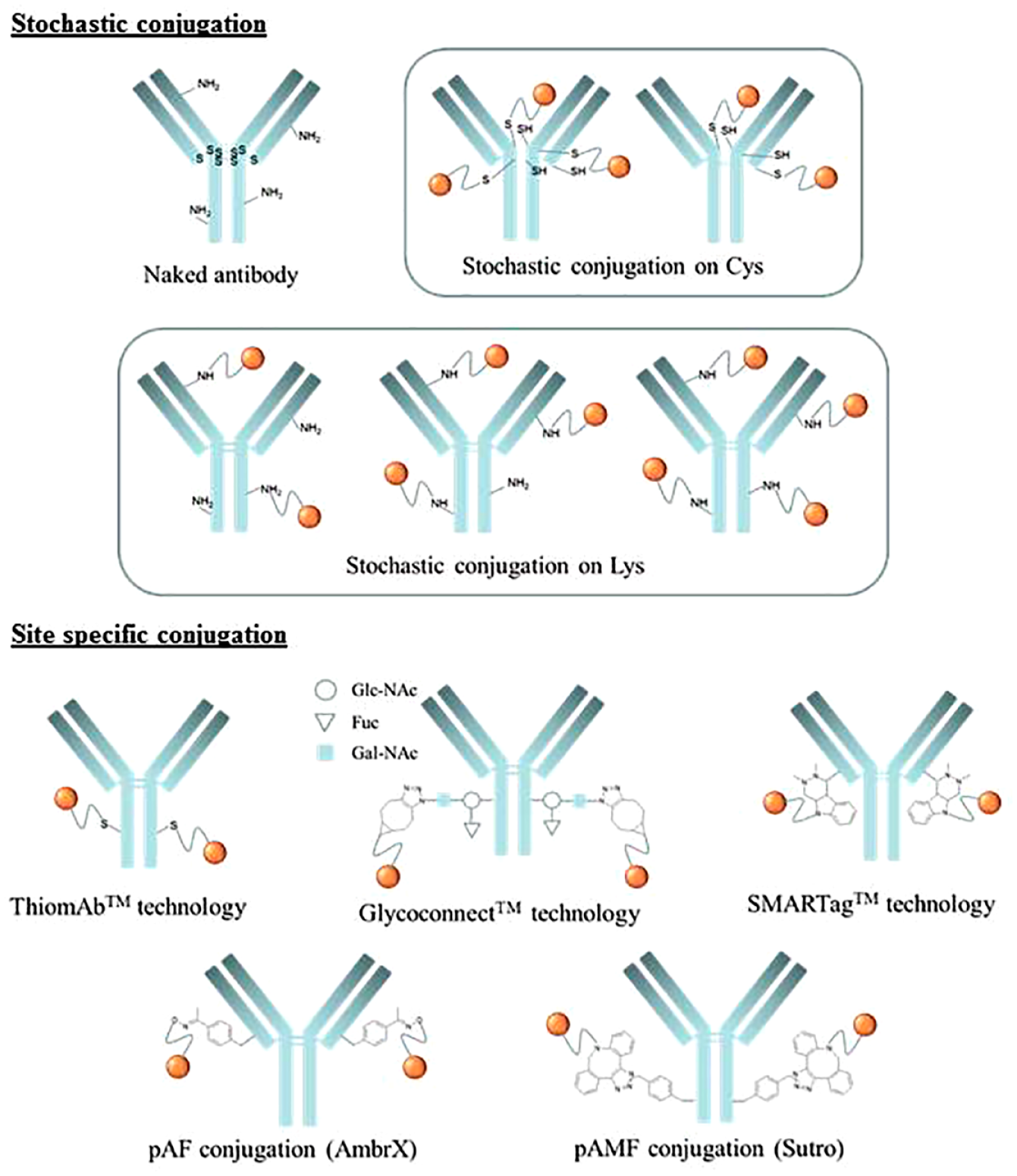
Conjugation technologies.
SSC on cysteines has been the most widely investigated method. Antibodies engineered to introduce cysteine residues in positions that are different from native interchain disulfides have enabled known and validated maleimide conjugation chemistry to be conducted and existing payloads to be exploited. Limited modification of the conjugation process is needed to ensure maintenance of interchain disulfide bonds and to avoid retro-Michael deconjugation. 144 Different methodologies have been developed to enable the precise introduction of reactive cysteines into antibodies. One such method is THIOMAB technology. This methodology was developed by Genentech, Inc. and led to several site-specific conjugated ADCs currently in clinical trials. The first THIOMAB, described in 2008,145,146 resulted from phage display screening of reactive cysteines on the light and heavy chains of the constant domains of the antibody Fab region of trastuzumab. The anti-MUC16-targeted antibody, generated using the THIOMAB–drug conjugate (TDC) methodology, demonstrated an improved therapeutic index. The HC-A114C variant emerged from this study as a valuable position for the production of MMAE TDCs and for the conjugation of batansine. 97 It also led to the first THIOMAB–antibiotic conjugate, DSTA4637S, currently in phase I. 147 TDC demonstrated noticeable, albeit minor, impact on tissue distribution 148 and revealed the importance of the chemical and structural dynamics of the conjugation site through comparison of the HC-A114C and LC-V205C positions for conjugation. The position of the conjugation site modulates the in vivo stability and therapeutic activity of TDCs. 144
The NexMab technology, consisting of introducing peptide motifs containing cysteine residues at the C-terminus of an antibody heavy chain, was also developed for alternative Cys SSC of MMAE. 149 Cysteine engineering was also applied to payloads with other mechanisms of action, with each agent having its optimal position. HC-S239C mutation was optimized to develop several PBD Talirine ADCs.108,109,150 Alternative methods for SSC on cysteine were also developed and applied to Tesirine. These were (1) a cysteine being inserted one amino acid before or after the three previous selected sites (HC-A114C, LC-V205C, and HC-S239C) 151 for conjugation; and (2) an antibody being engineered to LC-C214S, HC-C226V, and HC-C229V, leaving the native interchain disulfide position C220 as the only conjugatable cysteine, 116 and therefore enabling SSC on this position. In parallel, a cysteine residue was also engineered at the 442 position of the CH3 domain of the γ1 constant region of a CD123 targeting antibody to allow for SSC of the IGN payload DGN549. 122
More recently, de novo conjugation methods have emerged,
19
and the most advanced have led to compounds that are currently in phase I (
Whatever the SSC technology used, ADCs resulting from careful selection of an SSC site—some SSC sites work far worse than stochastic conjugates—demonstrated either an improved tolerability in rodents and nonhuman primates or an increased efficacy in mice versus stochastic ADCs. Some of these include ADCs that are still at the preclinical stage. 159 Strict head-to-head comparisons can be made in the case of Cys conjugation, maintaining all other properties the same (i.e., linkage chemistry, and linker and payload active metabolites). The superiority of the optimized site-specific Cys ADCs versus stochastic Cys ADCs comes only from conjugation site specificity. Direct comparison of stochastic conjugation to SSC is difficult in other cases due to different linkage chemistries, and occasionally different linkers. These linkers impact the nature of active metabolites that are released inside the cell following ADC processing, especially in the case of noncleavable linkers, and this is why the benefit of conjugation site specificity is difficult to evaluate in a systematic head-to-head comparison of SSC technologies. However, SSC enables alternative methods for optimization to be achieved through altering the tethered agent.
Optimization of FcγR Binding Properties of Antibodies
Depending on both the characteristics of the antibody target and the inherent properties of the cytotoxic payload of the ADC, Fcγ receptor (FcγR) binding properties of antibodies are important factors in determining the off-target toxicity of ADCs, such as, for example, neutropenic and thrombocytopenic adverse events. Several hypotheses have been proposed to explain the off-target toxicity of ADCs, one of them relating to the uptake and processing of ADCs by FcγR-expressing cells. Conversely, it has been suggested that FcγR-mediated effector functions may contribute to the clinical efficacy of ADCs. 142
The efficacy of Kadcyla is in part due to trastuzumab’s capacity to induce ADCC, 160 and it has been suggested that brentuximab’s capacity to cause antibody-dependent cellular phagocytosis (ADCP) in vivo is partly responsible for the efficacy of Adcetris. 161 Moreover, it has also been suggested that tumor-associated macrophage-processed MMAE-ADCs can mediate additional “bystander killing” of tumor cells by inducing ADC processing through FcγR interactions. 162 The observed thrombocytopenia side effect induced by Kadcyla is hypothesized to occur as a result of FcγRIIa-mediated internalization of the ADC into megakaryocytes, 163 resulting in the intracellular release of DM1. To avoid IgG1-mediated effector functions, IgG4 antibodies are used in Mylotarg and Besponsa. The choice of which IgG format to use in ADCs (e.g., IgG1 vs IgG4) may, therefore, define whether to introduce additional efficacy drivers in addition to the payload activity itself, 142 or to minimize side effects.
FcγR interactions between the ADC and the immune cells are important to manage. Because these interactions depend on the Fc isotype of the ADC, Fc engineering could also be exploited to modulate these interactions, particularly when the IgG1 format is used. 164 A method of impairing the FcγR binding is to use aglycosylated antibodies, which lack the Fc effector function, either through a single point mutation, such as N297A or N297Q, or through antibody production in a cell-free protein expression system lacking the glycosylation machinery, such as for STRO-001. 157 Other point mutations could also be used to reduce FcγR binding, as has been demonstrated for MEDI4276, where a L234F point mutation was combined with a S239C mutation for cysteine conjugation in order to reduce ADCC. 165 Finally, as conjugating payloads on the Fc region may impact FcγR binding, some SSC technologies use glycans present on the Fc portion of the mAb as payload attachment sites. The GlycoConnect technology was developed as a way to reduce off-target cellular uptake by FcγR-expressing cells 152 via enzymatic modification and conjugation of N-glycan residues at asparagine-297. More generally, conjugation in these antibody regions may have an impact on effector functions. Less advanced methods are currently under preclinical evaluation, such as conjugation after periodate oxidation of oligosaccharide structures at the fucose residue, 166 conjugation on thiofucose-engineered mAbs, 167 conjugation after remodeling of the glycans of antibodies with azido-containing sialic acid, 168 or conjugation on glutamines at positions 295 ± 297 of aglycosylated mAbs via transglutaminase.169–172
In contrast to the aim of reducing activity at the FcγR, conjugations that increase the binding affinity of the Fc domain to FcγRIIIa expressed on effector cells have been sought to enhance the ADCC activity of ADCs. To this end, stochastic conjugation on Cys residues has been used on afucosylated mAbs for GSK2857916 67 and BAT8003, 97 currently in clinical trials.
Innovation Related to Format
Modified Antibody Formats
Some modifications of mAbs in the Fab region are essentially aimed at optimizing tumor targeting and retention of ADCs in order to reinforce the selectivity and avidity toward the surface antigen, and to maximize ADC intracellular processing from internalization to trafficking to lysosomes. ADCs resulting from some of these technologies have already reached clinical trials.
The most advanced technology relies on biparatopic antibodies. This methodology was applied to HER2-directed ADCs. As HER2 is recycled back to the plasma membrane following spontaneous endocytosis, only a small fraction of Kadcyla is routed to the lysosome, where its active metabolite is released. Thus, it is likely that improving ADC internalization and lysosomal trafficking would significantly enhance HER2-directed ADC efficacy. Biparatopic antibodies targeting two nonoverlapping epitopes on HER2, with one arm binding to the same domain as trastuzumab, were developed. Indeed, these antibodies can induce HER2 receptor clustering, which in turn promotes robust internalization and strong lysosomal trafficking. 98 Exploiting this finding, ZW25, the antibody component of ZW49, 173 has been designed to interact with both the trastuzumab and pertuzumab binding domains, 174 already used in combination for treating HER2-positive metastatic breast cancer. 175 The development of the previous biparatopic HER2-directed ADC MEDI4276 was discontinued after phase I completion due to payload-related toxicities. This ADC was designed to combine binding to the same domain as trastuzumab, as well as another unique epitope on HER2 that was different from pertuzumab.98,176 For similar reasons, the idea of a bispecific antibody targeting both the tumor-specific antigen HER2 and the cell surface antigen APLP2 or PRLR has emerged, but this approach has yet to demonstrate broad utility.177–179
Another approach aims at reducing nonspecific uptake of ADC as a result of conditional activation of the conjugate at the tumor site, through increased selective ADC binding on tumor cells versus normal tissues. This approach could also enlarge the druggable target space of ADCs when normal tissue expresses the target receptor, in addition to the high level expressed on tumors. One technology takes advantage of the tumor microenvironment (TME) to localize treatment to the tumor more effectively: target-binding regions of antibodies can be activated only in tumor tissue to ensure antitumor activity, while avoiding on-target toxicity in normal tissues. Exploiting this approach, Probody makes use of a N-terminal prodomain that masks the target-binding region of the antibody in circulation, which is removed through the action of proteases, such as matriptase, uPA, and legumain, that are active in the TME. 180 Two Probody drug conjugates are currently under evaluation: CX-2009 targeting CD66 (ALCAM) that is conjugated to SPDB-DM4, 82 and CX-2029 targeting CD71 (TFRC) conjugated to vc-PABC-MMAE. 54 More recently, Conditionally Active Biologics (CAB) have been developed as a new kind of antibodies that can bind to their target when in the vicinity of tumors and lose their affinity when less proximal. CAB were identified following library screening in differential conditions (e.g., binding at pH 6 and no binding at pH 7.4), with the objective of increasing selectivity toward tumor cells. Two CAB drug conjugates are currently in phase I/II: BA3011 targeting AXL 181 and BA3021 targeting ROR2. 182
Small Formats
Indications for approved ADCs are mostly for liquid tumors. ADCs targeting solid tumors have usually failed in clinical trials, with low tumor penetration of such large macromolecules being considered as the cause of their limited efficacy in vivo. As an alternative, developing cytotoxic conjugates with smaller targeting moieties has been explored as a strategy to cope with the size of ADCs.183,184 Different classes of novel entities are still at the preclinical stage, from antibody–fragment drug conjugates (e.g., Fab, diabody, and scFv) to small protein scaffold drug conjugates (e.g., Affibody, DARPin, Abdurin, and Adnectin),185,186 and a clear benefit from this kind of modality still needs to be demonstrated as superior tumor penetration is usually coupled with faster systemic clearance and subsequent limited in vivo efficacy. 179
To date, only three drug conjugates smaller than antibody–fragment drug conjugates are currently being evaluated in clinical trials, namely, peptide–drug conjugates PEN221
187
and BT1718
188
and a small molecule–drug conjugate PEN866
189
(
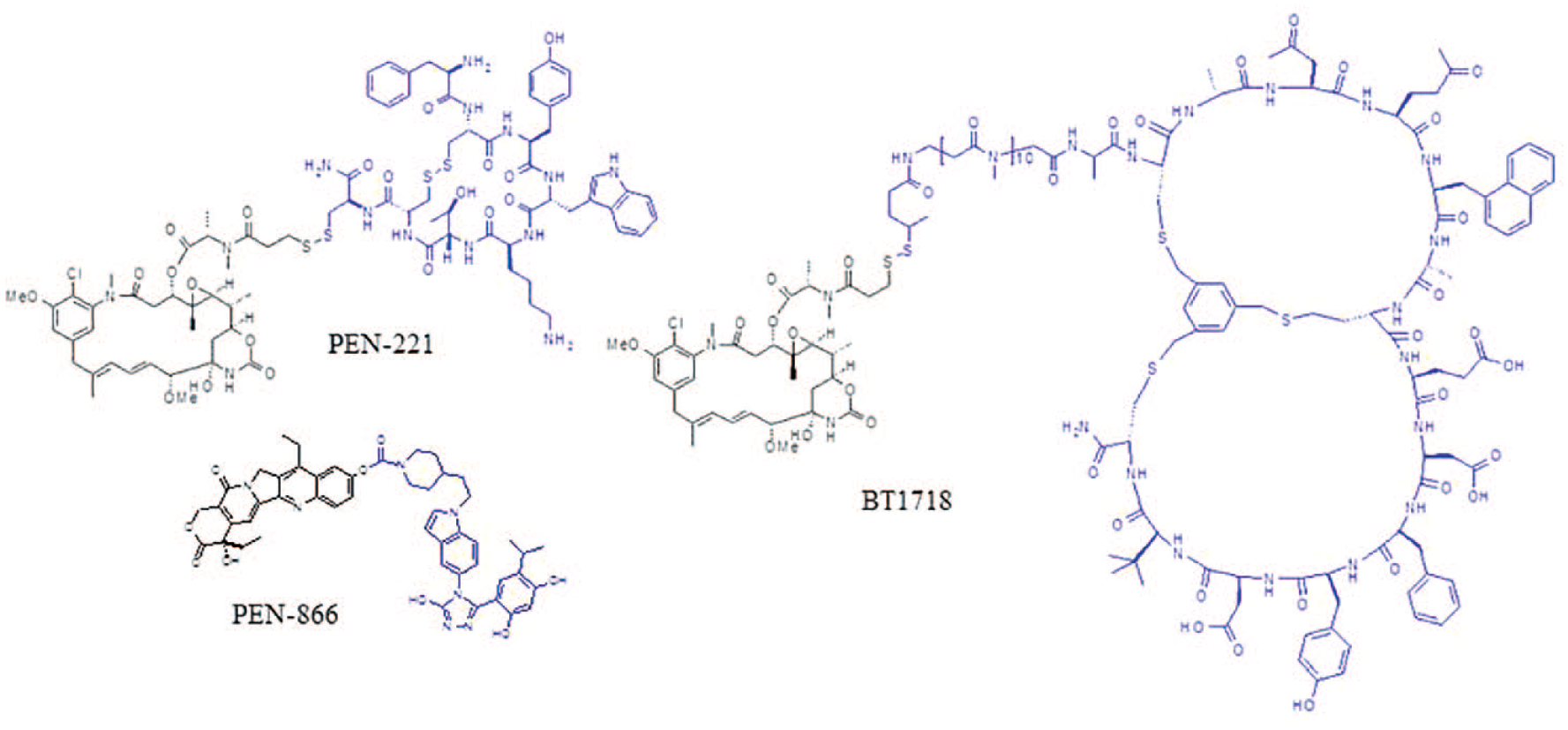
Small-format drug conjugates at the clinical stage.
Conclusion and Perspectives
If the story of the first marketed ADC, Mylotarg, was one of ups and downs, the stories of Adcetris and Kadcyla are ones of success, with global market sales for both compounds reaching US$1.87 billion in 2018, with forecasts for 2024 estimated to be US$4.29 billion. Moreover, the forthcoming years may see increased numbers of approvals in the ADC field, which has already begun with the recent approval of Besponsa, Polivy, Padcev, and Enhertu in 2017 and 2019. Recent approval of Enhertu validates topoisomerase I inhibitors as valuable components of future ADCs, with an expectation to enlarge clinical indications to other solid tumors. Looking to the future, four new ADCs are in a position for potential success, with two fast-track designations approved: mirvetuximab-soravtansine in platinum-resistant ovarian cancer and trastuzumab-duocarmazine in HER2+ breast cancer. Furthermore, two breakthrough designations for ADCs described in this review have been granted: sacituzumab-govitecan in triple-negative breast cancer and belantamab-mafodotin in multiple myeloma.
Trastuzumab-duocarmazine 191 is built on the same antibody as that of Kadcyla and Enhertu, sharing the same HER2+ breast cancer indication. The major difference between Kadcyla, Enhertu, and this other ADC is the payload consisting of a new DNA binder (duocarmycin based). Sacituzumab-govitecan 130 is another ADC based on topoisomerase I inhibitor SN-38. This ADC contains a TROP2-directed antibody. The future success of this product may validate the concept of using a moderately potent cytotoxic drug with slow release at the tumor site in addition to expected processing after internalization. Building upon optimization of already approved ADC payloads, mirvetuximab-soravtansine 87 is composed of an FRα-directed antibody linked to a sulfo-SPDB-DM4 payload. This payload was the result of the optimization of SMCC-DM1 as a cleavable version. Belantamab-mafodotin 67 is composed of a B-cell maturation antigen (BCMA)-directed antibody linked to mc-MMAF, a noncleavable auristatin developed as an alternative to the cleavable MMAE payload. In the case of this ADC, optimization also involved afucosylation of the antibody as a way to increase effector functions. Taken together, these achievements suggest positive signs after decades of slow progress in developing clinically effective ADCs. The number of positive outcomes may well increase in the coming years as a result of a better understanding of the criteria needed to attain efficacy at the preclinical stage, in addition to current innovations that may offer better cooperative links between the target, mAb, payload, and indication. Additionally, further improvements that have been described in this review and that are still at an early clinical stage (i.e., SSC and modified formats) offer promise for the future of this modality.
The growing promise of the use of immunotherapy to treat cancer could provide a boost to ADCs as a modality for use as therapeutic agents, due to three specific areas of interest. The first is the potential combination of ADC treatments with immunotherapy 192 following the observation that Kadcyla renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. 193 Evidence in in vivo preclinical models 194 suggests that there is therapeutic synergy when ADCs and immunotherapy are combined, and combinations of ADCs with anti-PD-1 antibodies are currently being explored in clinical trials. 195 The second option could be the use of immune-stimulant molecules as new payloads for ADCs and the first ADC of this type, NJH395, is already in phase I. This innovative ADC comprises an anti-HER2 antibody conjugated to a TLR7 ligand, where the immune-stimulating moiety is aimed at enhancing the immune-mediated killing of HER2-expressing tumor cells, by activating dendritic cells within the tumor, through the stimulation of toll-like receptors present on immune cells. In addition, targeting receptors present on immune cells for the delivery of cytotoxic or immunomodulatory molecules through ADC modality could also be a field of interest, and first conjugates of that type are emerging. 196 Of note, the tumor-supportive macrophage population is an attractive target for the treatment of macrophage-rich tumors. Indeed, macrophages could be reprogrammed/repolarized to a state where they suppress rather than stimulate tumor growth. This strategy is being explored by an antibody targeting CD163, an effective internalizing receptor, highly expressed in the immunosuppressive macrophage population, conjugated to dexamethasone, a glucocorticoid anti-inflammatory drug. 197
The ADC field may finally be in a position to open the way to a real breakthrough in the fight against cancer. Moreover, some other disease areas could also emerge as new fields of interest for this therapeutic modality. Indeed, some ADCs have already reached phase I for the treatment of rheumatoid arthritis (ABBV-3373) and infectious disease (DSTA4637S 147 ), and glucorticoïds are being considered as payloads for the treatment of lupus erythematosus. 198 Even if it is too early to tell whether these ADCs will be future clinical successes, these innovations could also enlarge the scope of future ADCs.
Footnotes
Acknowledgements
The authors thank Brigitte Demers and Vincent Mikol for critical reading of the manuscript, Stéphane Cheradame for competitive landscape documentation, and Kwame Amaning for help in proofreading.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.