
Abstract
Identifying the cell surface receptors of natural ligands, and deconvoluting the receptor targets of candidate drug leads, presents a challenge in medical research and drug discovery. Traditionally, success rates have been low, and screening efforts often generate numerous false-positive hits that require extensive follow-up to either validate or disregard. If successful, receptor identification enables the discovery of previously unknown, disease-relevant targets, provides critical insights into biological pathways and disease processes, and allows for secondary targets to be uncovered. By expressing the majority of the human plasma membrane proteome in human cells on glass slides in situ, human cell microarray technology provides a powerful approach for identifying receptor target interactions. This approach significantly increases the success rates in identifying specific primary receptor targets and off-targets while limiting the number of false-positive hits. Here we describe cell microarray technology, focusing on new advances including the use of whole cells as bait for receptor interactions, and the inclusion of secreted proteins that widens the utility of the technology in off-target screening.
Introduction
In medical research and drug discovery, it is often highly advantageous—or even essential—to identify the cell surface proteins that are targeted by a molecule of interest. Broadly, this falls within three categories: identifying receptors for natural ligands, elucidating the targets of molecules with a desirable function or phenotype but with an unknown target, and assessing the “off-target” liabilities of promising drug leads.
First, understanding the cell surface interactions of naturally occurring ligands will shed light on normal biological processes as well as help to elucidate the mechanism of disease. In this respect, receptor identification is relevant in countless, diverse areas ranging from linking a known immune checkpoint protein with its receptor binding partner, to identifying the human receptors that mediate bacterial, viral, or parasitic infections. Knowledge of key receptors implicated in the immune response, or in the progression of a disease, provides researchers with new opportunities for the development of targeted therapies.
Second, deconvoluting the receptor targets of promising molecules, including antibodies and small molecules, that have been selected through functional or phenotypic screening is a valuable step in phenotypic drug discovery. Despite its importance, target deconvolution is often approached with low expectations of success. As the functional target for all phenotypic antibodies (as well as some small molecules) will be a cell surface protein, plasma membrane proteins are a priority focus in the development of target deconvolution approaches.
Third, in drug development—even when the primary drug target is already known—it is critical to understand if a drug candidate interacts with additional proteins, and what the identities of those secondary targets, or off-targets, are. Off-target interactions can have a significant impact on safety or toxicology and can unfavorably affect pharmacokinetics. Knowledge of off-target liability at the late preclinical stage, or even earlier during lead selection, can better inform the risk–benefit analysis, potentially reducing late-stage failures, which could carry significant financial benefits for sponsors.
Despite this clear need for effective receptor identification systems, traditional approaches for identifying receptor targets for natural or phenotypic molecules have had very low rates of success. Furthermore, even if a candidate target protein is identified using a traditional protein array or receptor pull-down assay, its significance as a primary receptor may be masked by a multitude of false-positive interactions. This can lead to substantial follow-on hit validation projects in order to reveal any meaningful results. As a consequence of high false-negative and high false-positive rates, both ligand de-orphanization and target deconvolution often represent bottlenecks in research programs. Even fairly recently, when faced with seemingly intractable cases, key opinion leaders in phenotypic development have weighed the relative risks of approaching regulators with a clearly functional drug but without a thorough understanding of its mechanism of action, versus completely halting the development of a promising candidate due to the inability to pinpoint its primary receptor target. 1
The limitations of the traditional technologies have largely been attributed to the nonnatural environment in which receptor proteins are presented. Proteins arrays, which serve as rapid and relatively low-cost screens, involve probing libraries of membrane proteins (and other proteins) that have been spotted onto slides with a ligand of interest in order to identify binding events. However, as the spotted proteins are not always full length and are not presented in a physiological conformation within the cell membrane, there can be a high rate of false negatives, whereby real interactions are not detected. Additionally, a high incidence of false-positive hits makes interpreting the data from protein array screens difficult and time-consuming. Although traditional protein array screening may be a useful tool, overall confidence in the results can be very low.
In contrast, native cDNA expression systems allow for biologically relevant interactions occurring at the surface of living cells to be examined at an individual, specific protein level. By comparison with more precedented arrayed protein techniques, the human cell microarray format confers significant advantages, principally the enabling of membrane protein overexpression and presentation in a mammalian cellular context. 2 This allows each protein to be properly folded, glycosylated, and potentially multimerized prior to screening with the test ligand. This increases the chances of capturing “real-life” interactions and has even detected interactions mediated or directly influenced by specific posttranslational modifications. 3 As the identities of the arrayed expressed proteins are known in advance, results can be very quickly determined via positional mapping, rather than requiring mass spectrometry analysis. Low-affinity interactions (i.e., <1 µM) can be readily detected under normal screening conditions, 2 with signal amplification further boosting the detection of very low-affinity interactions, if required.
This article describes the human cell microarray technology, providing examples of applications from published studies. Two new, powerful advances that provide further advantages beyond standard protein array screening are then described. These are the ability to identify receptor–receptor interactions using whole-cell “baits” instead of soluble ligands, and the incorporation of secreted proteins into the assessment of off-target liability.
Human Cell Microarray Methodology
Human cell microarray technology involves the arraying of comprehensive libraries of cDNA expression vectors on slides, overexpressing the encoded proteins in human embryonic kidney-293 (HEK293) cells, and screening for interactions of a test molecule against all cell-expressed proteins. Each cDNA clone within the library encodes an unmodified, untagged human protein from a carefully collated library of plasma membrane protein targets. The human cell expression system provides a more natural environment for normal protein trafficking, folding, posttranslation modifications, and localization at the cell surface.
The platform technology is compatible with a vast range of test ligands including human and nonhuman antibodies,4,5 single-chain variable fragments (ScFvs), 6 proteins, peptides, and small molecules. 2 Target receptors for more complex ligands can also be readily identified. For example, human receptors targeted by viruses 7 and parasitic infections3,8 have been elucidated using the system, with these discoveries widening the scope for the development of vaccines and new therapies.
All molecules to be tested using the cell microarray approach are initially screened for background binding to nontransfected human HEK293 cells. This is to ensure that there is minimal or insignificant binding to endogenously expressed human proteins and to optimize the dose at which to screen each test ligand. The technology was optimized using HEK293 cells as these cells are a human, adherent, readily transfectable cell line.
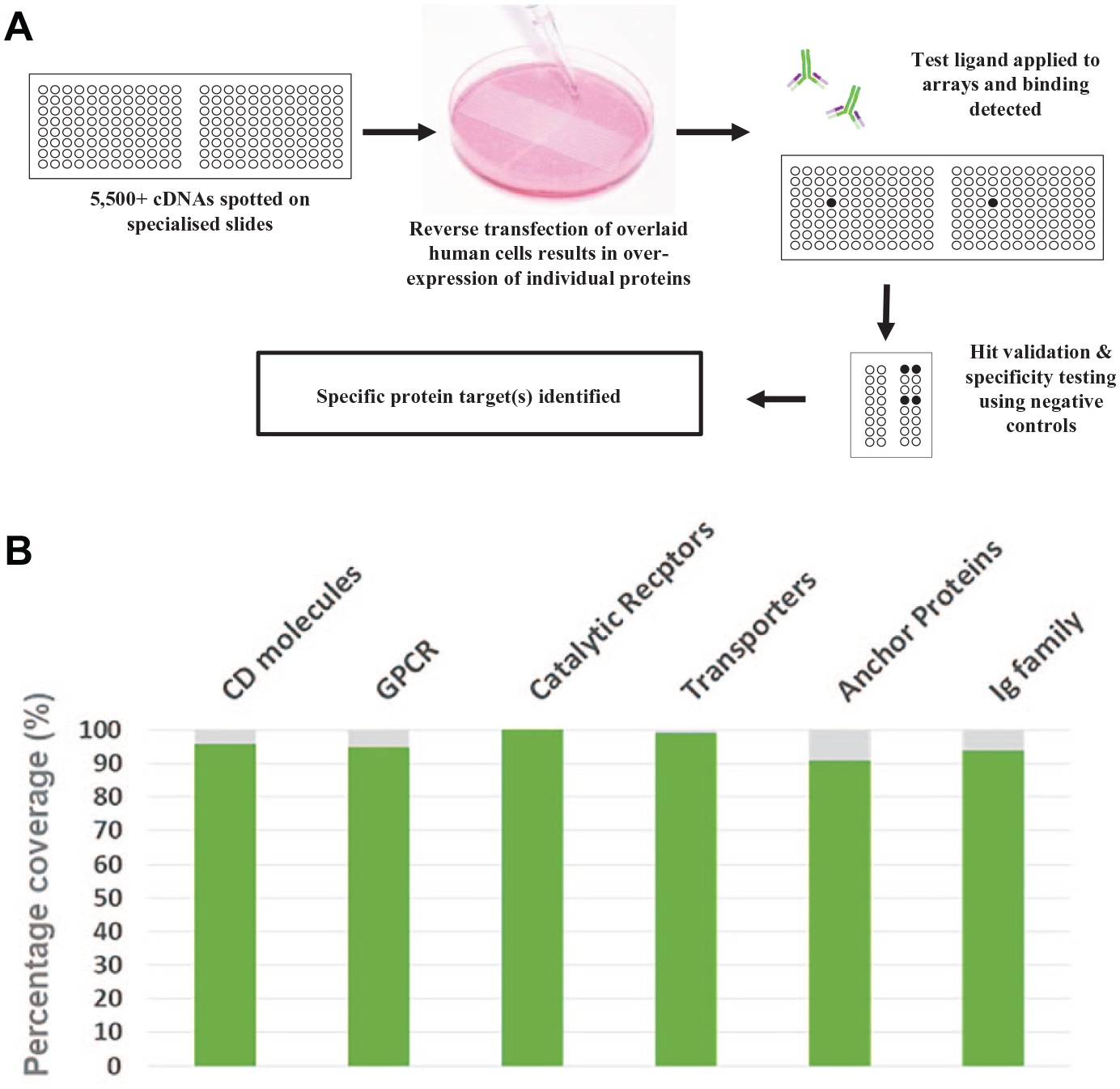
Thousands of cDNA expression vectors encoding individual human proteins are then arrayed, in duplicate, onto specialized slides. Each expression vector also encodes ZsGreen1, a green fluorescent protein, which is used to positionally locate subsequent positive interactions, and to ensure that a minimal threshold of transfection efficiency has been achieved or exceeded on every slide. HEK293 cells are seeded on the arrays and become reverse transfected, resulting in distinct clusters of cells (“spots”) that overexpress the individual protein encoded by the cDNA below. The test molecule is then added to the cell microarrays, and screening can be performed on live or fixed transfected cells. Counterintuitively, fixed cell screening often results in stronger binding signals on the microarrays, indicating that this is a more sensitive approach, rarely with a detrimental effect on the target epitope. An overview of the technology is provided in Figure 1A .
(
Interactions between the test molecule and its overexpressed receptor are detected by monitoring gain of binding via a specific detection system (e.g., a fluorescently labeled antibody to the test molecule, or to an integral tag such as Fc, biotin, or FLAG). “Hits” from a screen against the entire library are analyzed and identified, using the co-expression of ZsGreen1 to define their precise location on the slides.
Clones encoding these hits are reexpressed in duplicate, along with appropriate positive and negative control receptors, and a second round of screening is performed with the test molecule, and negative control treatments, in order to test the reproducibility of the identified interaction(s), and to assess specificity. Following this confirmation screen, further validation using live transfected cells and a flow cytometry-based readout may also be carried out, if required. This provides additional validation as well as allowing for titration experiments whereby increasing doses of the test molecule can show a curve shift against each receptor produced by varying degrees of molecule–receptor binding. Typically, only one or two highly specific hits are returned with a standard cell microarray screen and the overall false-positive rate is very low.
Development of a Robust Membrane Protein Library
The original goal in the development of the cell microarray technology was maximal coverage of the human plasma membrane proteome. However, more than 10 years of library development and testing of the platform with thousands of molecules has resulted in a more refined approach, rather than default inclusion based on simple categorization. Rigorous analysis of the bioinformatic and published evidence behind a protein’s classification as a plasma membrane protein has led to careful exclusion of miscategorized proteins and produced a “high confidence” library for screening. Obviously, proteins with strong evidence of an extracellular domain (ECD) ranked particularly high when selecting candidates. However, there has been no selection bias toward any protein family. This has resulted in good representation across all subclasses of membrane protein families ( Fig. 1B ). Many proteins have multiple isoforms represented within the library; however, the canonical or longest isoforms have always been prioritized. This cautious approach has resulted in a robust library that now includes >80% of these high-confidence plasma membrane proteins and provides a very good chance of identifying interacting proteins.
Case Examples: Identifying Immunotherapy Targets Using Cell Microarray Screening
Extensive validation of the cell microarray technology has been carried out using known ligand–receptor pairs and through hundreds of screening projects that have been carried out on behalf of drug sponsors and academic research groups. The following examples briefly outline two published cases where novel targets were identified in the development of cancer therapeutics, including previously unidentified and clinically relevant off-targets.
Case 1: Discovery of Siglec-6 as a CLL Target
Isolating the antibodies that are involved in the human immune response to cancer by selecting and enriching those that are abundant in patients that have displayed a good response to anticancer treatment can result in the identification of novel and important anticancer receptor targets. This approach has recently been used to discover potential new treatment options for chronic lymphocytic leukemia (CLL)—the most common form of leukemia in the United States. 9 Currently, CLL can only by cured by allogeneic hematopoietic stem cell transplantation (alloHSCT). Although this stem cell transplantation is associated with high morbidity and mortality, it can be effective, most likely due to posttransplantation antibodies that are specific to CLL antigens promoting an immune response to the disease. Chang et al. 9 isolated and enriched post-alloHSCT antibodies, which they then screened for target receptor interactions using various techniques. Attempts to uncover the antigen by immunoprecipitation and mass spectrometry proved unsuccessful. However, cell microarray screening identified Siglec-6 as a strong specific interaction, which was subsequently verified by the group to be a CLL-specific antigen. The study not only delivered fully human, antihuman endogenous Siglec-6 monoclonal antibodies—that proved to be safe in early studies—but also presented Siglec-6 as a promising new target for the development of a novel immunotherapy to help patients with CLL.
Case 2: Is There Only One Target?
Unless evidence presents to the contrary, the “monospecificity” of a therapeutic antibody is often assumed. This second published study highlights the pitfalls of assuming that only a single target is responsible for the mechanism of action of a therapeutic molecule, noting that polyspecificity is a significant risk factor in the successful development of antibody-based drugs.
The anti-PD1 antibody SHR-1210, also known as camrelizumab, was selected for the study. 10 Despite displaying the expected biological activity in early clinical studies for solid tumors, the toxicity profile of camrelizumab was notably different from that of other anti-PD1 immunotherapies. The majority of patients (>80%) receiving camrelizumab have developed treatment-related capillary hemangioma, a benign tumor formed by a collection of excess blood vessels. 11 This led to the hypothesis that the target binding domains of SHR-1210 might interact with previously unidentified and unpredictable receptors that are associated with vascular development or tissue differentiation. Cell microarray technology was used to investigate the plasma membrane protein targets of SHR-1210-IgG1. This rapidly uncovered three human receptor interactions, in addition to PD1 binding, that may explain the molecular mechanisms behind the observed toxicity. These interacting receptors—VEGFR2, FZD5, and ULBP2—were confirmed using multiple orthogonal approaches.
Notably, off-target binding to VEGFR2 and possibly frizzled class receptor 5 (FZD5) may stimulate vascular neogenesis and lead to the development of hemangioma. Importantly, the off-target binding of VEGFR2 actually led to potent activation of that receptor, which is known to be a key signaling event that stimulates capillary blood vessel proliferation.
The discovery that the target binding domains of camrelizumab exhibit relevant off-target reactivity led to further investigations that confirmed that the antibody’s polyspecificity existed prior to humanization. Subsequent engineering of the antibody through mutagenesis and reselection generated novel antibodies that targeted PD1 and showed improved pharmacological properties, but which did not bind to the three off-targets identified for the original antibody. This study shows that clinically relevant polyspecificity is a phenomenon that exists, but that it can be identified and rapidly engineered out of a therapeutic protein. This can potentially lead to improved potency and reduced toxicity in a final therapeutic product. 10 Early screening to identify any potential cross-reactivity can have major safety implications for trial participants and patients as well as help to direct clinical resources toward drug candidates that are more likely to succeed.
Results and Discussion
New Developments in Cell Microarray-Based Screening
The previous examples show the utility of cell microarray screening of ligands (such as proteins, antibodies, and peptides) to identify plasma membrane binding partners. However, two recent advances now allow the technology to be used to identify ligand–receptor interactions using whole-cell baits instead of just soluble molecules, and also to screen for interactions with proteins that are usually secreted from the cell.
Whole Cells as Bait
The standard approach to uncover receptor–receptor interactions, such as immune checkpoint interactions, using cell microarray technology is to screen a soluble version of the test receptor. Often, this is a tagged ECD domain fragment of the test receptor, linked to a tag, such as an Fc domain. However, an alternative and attractive approach is to screen whole cells expressing the receptor as bait against the cell microarray slides. The test protein will be present in its full form, rather than just available as an isolated fragment (e.g., an ECD), and will be expressed in its natural membrane context. So-called bait cells could contain endogenously expressed test protein, or the cells could be transfected with test protein. This is useful for screening proteins that have been difficult to isolate and provides an alternative strategy if there has been limited success in receptor identification efforts using a soluble form of the ligand.
Bait cells are labeled with a fluorescent dye and then incubated on the HEK293 cell microarrays at a specific cell density that is optimized for each different cell type screened. Following a short incubation, a careful washing step is required in order to effectively remove unbound cells while not disrupting the cells that are bound to their specific counterreceptor(s) expressed by the HEK293 cells. As the cells are labeled, the test protein remains untagged and unaltered. As such, it should be correctly folded and presented at the cell surface in the context of any additional accessory proteins (if required). As each bait cell will express multiple copies of the ligand, it will bind to the cell microarrays via multiple ligand–receptor interactions. This may provide an avidity gain over a single soluble ligand–receptor interaction. For all of these reasons, screening whole cells is potentially a much more sensitive approach for identifying receptors.
As with soluble test proteins, putative receptors of the bait cells are identified via analysis of fluorescence. As whole cells are being used as the bait, the screen will also identify receptor–receptor interactions that are unrelated to the test protein of interest. In order to distinguish the specific test protein–receptor interaction from the other cell–cell interactions, it is necessary to conduct a counterscreen using cells that are test receptor negative. For stable transfectants that are overexpressing the test protein, parental cells are an ideal negative control. For endogenously expressed ligands, a knockout or knockdown cell line would be required to differentiate between the whole-cell binders and the ligand-specific interactions.
This whole-cell bait approach is now being utilized for receptor identification, notably in uncovering immune checkpoint interactions for ligands that have been difficult to de-orphanize. Figure 2 shows the proof-of-principle data assessing the feasibility of the technique using transfected Jurkat cells, an immortalized T-cell line. Jurkat cells that overexpressed the PD1 ligand (bait cells) bound to HEK293 cells overexpressing PD-L1, as well as a suite of known T-cell interactors. In contrast, parental Jurkat cells showed binding to the known T-cell interactors, but not to PD-L1 (not shown). This validation of the whole-cell binding approach with a known immune checkpoint pairing exemplifies the value of this technique as another tool for uncovering novel receptor–receptor interactions.
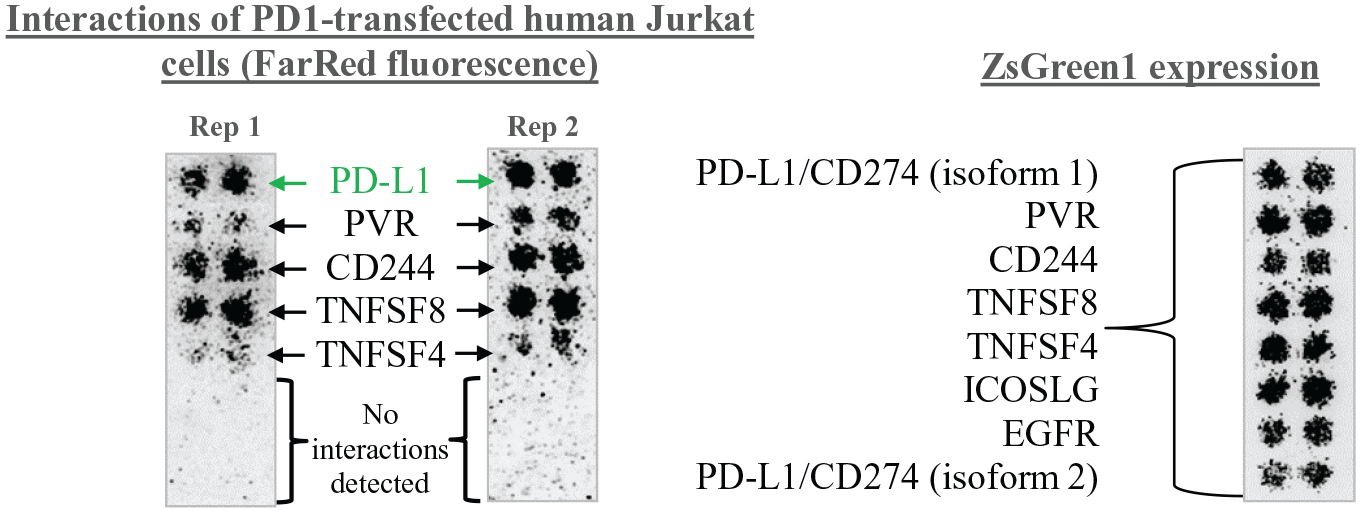
PD1-transfected Jurkat cells were labeled with a FarRed fluorescent cell dye and added at a 4:1 molar ratio for 1 h to slides of fixed untransfected HEK293 cells, and HEK293 cells overexpressing PD-L1/CD274 (two isoforms), four known T-cell interactors (PVR, CD244, TNFSF8, and TNFSF4), and two negative control receptors (ICOSLG and EGFR). ZsGreen1 expression from each spotted expression vector is shown on the right-hand side. The PD1-transfected Jurkat cells, but not the parental Jurkat cells, showed strong binding to PD-L1 isoform 1 but not to PD-L1 isoform 2. As expected, the Jurkat cells also showed binding to the known Jurkat interacting proteins PVR, CD244, TNFSF8, and TNFSF4, but not to ICOSLG or EGFR (left-hand side).
Cell microarray technology is the only platform for screening whole-cell baits for binding to cellular overexpression libraries. As such, it has become integral to off-target safety screening of chimeric antigen receptor (CAR) T cells.12,13 Minimizing the risk of triggering an inappropriate immune response through unanticipated off-target binding of engineered CAR T cells is essential in ensuring patient safety. Precursor antibodies or ScFvs, as well as the final engineered CAR T cells, can all be screened for off-targets, and data from these studies are being used in regulatory submissions, including the biologics license application (BLA) for Novartis’s tisagenlecleucel (Kymriah) in the United States, 14 Europe, 15 and Japan. 16
Incorporating Secreted Proteins
The cell microarray technology is widely used to screen biotherapeutic molecules for off-target binding to human cell surface proteins expressed in situ. However, binding to secreted proteins can also pose a potential toxicology risk and impact on the pharmacokinetic profile of a drug. 17 As secreted proteins are not naturally localized on the cell surface, expression of these proteins using the standard cell microarray procedure would result in release of the protein from the arrays and interactions would most likely go undetected. As such, adaptation of the technology has been necessary in order to incorporate secreted proteins.
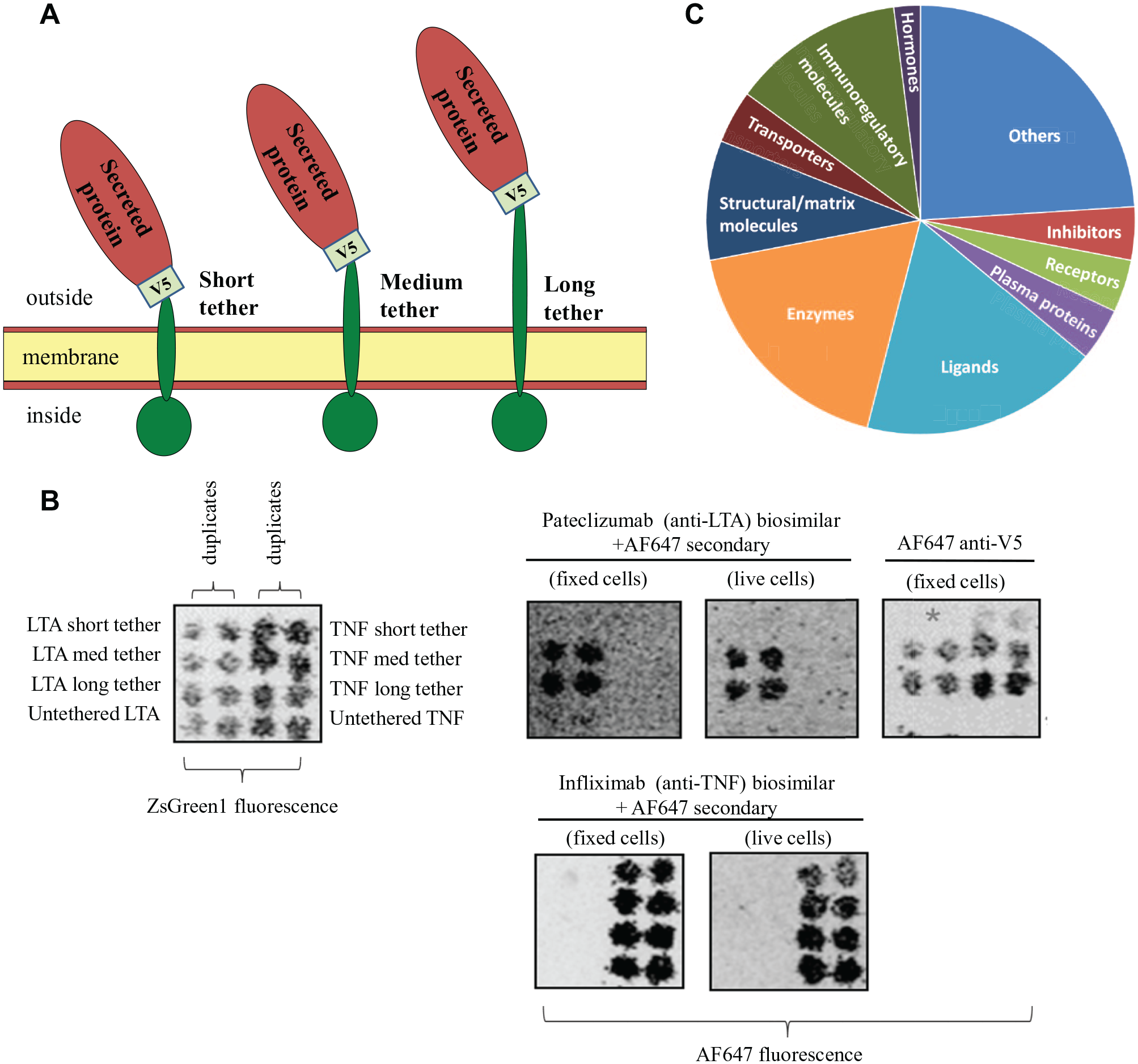
Novel constructs that fuse the proteins to inert transmembrane and intracellular domains have been deployed to allow for expression and binding analyses of “secreted” proteins using the platform. Three lengths of tether were tested—short, medium, and long ( Fig. 3A ). A V5-tag was also incorporated, allowing expression of the secreted protein to be verified and semiquantitated. Co-transfected ZsGreen1 (a green fluorescent protein) allows for transfected cell clusters to be mapped and for interacting proteins to be identified by their position on the arrays.
(
Multiple antibody–secreted target pairs were used to determine optimal construct length and establish proof of principle. Data are shown for a pateclizumab biosimilar against its secreted target lymphotoxin-alpha (LTA), also known as TNF-beta ( Fig. 3B ). Transfection efficiency and spot positions were confirmed using ZsGreen fluorescence (left-hand panel). Pateclizumab showed binding to the medium- and long-tethered forms of LTA, but not to untethered LTA or the short-tethered form of LTA. Pateclizumab showed no cross-reactivity against the related family member TNF (a transmembrane protein), despite confirming its expression by the binding of infliximab. The proof-of-principle results on a subset of secreted targets led to the selection of long-tethered constructs for the development of the full secreted library.
In total, approximately 2000 candidate secreted proteins have been identified and prioritized for inclusion based on manual curation of subcellular location, the presence of a signal peptide sequence, and/or prediction algorithms. Toxicology and bioinformatics teams from four major pharmaceutical companies have also advised in order to prioritize key secreted proteins that are particularly relevant to off-target screening. Currently, a library of 1400 of these high-confidence proteins has been incorporated into the cell microarray platform, with the remaining 600 under development ( Fig. 3C ). These future developments will include membrane proteins that have secreted isoforms as well as natural proteolytic fragments of membrane and secreted proteins (e.g., fragments of complement proteins).
This expansion to the cell microarray platform has extended target profiling beyond natural plasma membrane targets and is particularly beneficial in the assessment of secondary, off-target activity that has the potential to cause clinical toxicity of a biotherapeutic in development.
Impact
The examples described here provide an introduction to some of the typical applications of the cell microarray technology as well as discussing the recent advances in screening whole cells and assessing secreted protein targets. In cases where there is no prior knowledge of a protein target, valuable intellectual property can be generated as a result of a cell microarray screen. Screening results have contributed to patent applications, including finding the binding partner for a novel immune checkpoint receptor within the TIGIT family 18 and identifying a putative VISTA receptor. 19 Although the technology was initially developed for ligand de-orphanization and target deconvolution, due to its very low rate of false positives, its use in off-target assessment is now widespread in biotherapeutic drug development, including antibody-based molecules and CAR T cells. It has been noted by Comess et al. that using cell microarray technology, “either a valid target is identified, or no targets are identified at all, which contrasts with the broad swath of disproven targets found in every other target identification method.” 2
Although the technology is used with a wide range of ligand types, including proteins, cells, viruses, and small molecules, other modalities present new opportunities. For example, the development of an assay for specificity screening of engineered T-cell receptors (TCRs) would be incredibly beneficial to cell therapy developers who have limited options for preclinical safety assessment of off-target binding. As TCRs recognize fragments of an antigen as peptides bound to major histocompatibility complex (MHC) molecules, they are not compatible with cell microarray screening in its current form. Adaptation of the cell microarray technology to accommodate TCR screening is one development objective.
In conclusion, rapid and reliable receptor identification and target profiling plays a key role in driving progress in medical research and the development of novel therapies. 20 Continued extension of the technology within the plasma membrane proteome and further extension to the secretome, as well as to different ligand types, will continue to make the technology more valuable in accurately and efficiently profiling target binding.
Footnotes
Acknowledgements
The authors would like to thank Elizabeth Kingsley for assistance with preparing the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employed by Retrogenix Limited, and their research and authorship of this article was completed within the scope of their employment with Retrogenix Limited.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.