
Abstract
Nicotinamide mononucleotide adenylyltransferase (NMNAT; EC 2.7.7.1) catalyzes the reversible production of NAD+ from NMN+ and ATP and is a potential drug target for cancer and neurodegenerative diseases. A sensitive bioluminescent assay format suitable to high-throughput screening (HTS) and mechanistic follow-up has not been reported and is of value to identify new modulators of NMNATs. To this end, we report the development of a bioluminescent assay using Photinus pyralis ATP-dependent luciferase and luciferin for NMNAT1 in a 384-well plate format. We also report a mechanistic follow-up paradigm using this format to determine time dependence and competition with substrates. The assay and follow-up paradigm were used to screen 912 compounds from the National Cancer Institute (NCI) Mechanistic Diversity Set II and the Approved Oncology Set VI against NMNAT1. Twenty inhibitors with greater than 35% inhibition at 20 µM were identified. The follow-up studies showed that seven actives were time-dependent inhibitors of NMNAT1. 2,3-Dibromo-1,4-naphthoquinone was the most potent, time-dependent inhibitor with IC50 values of 0.76 and 0.26 µM for inhibition of the forward and reverse reactions of the enzyme, respectively, and was shown to be NMN and ATP competitive. The bioluminescent NMNAT assay and mechanistic-follow-up will be of use to identify new modulators of NAD biosynthesis.
Introduction
NAD and its phosphorylated form, NADP, are essential molecules to all organisms, acting as redox factors and substrates in many biological pathways. NAD biosynthesis fuels bioenergetic processes and maintains balanced cell regulation. Nicotinamide mononucleotide adenylyltransferase (NMNAT; EC 2.7.7.1) catalyzes the reversible reaction of nicotinamide mononucleotide (NMN+) or nicotinic acid mononucleotide (NaMN+) and ATP to form NAD+ or NaAD+ and pyrophosphate (
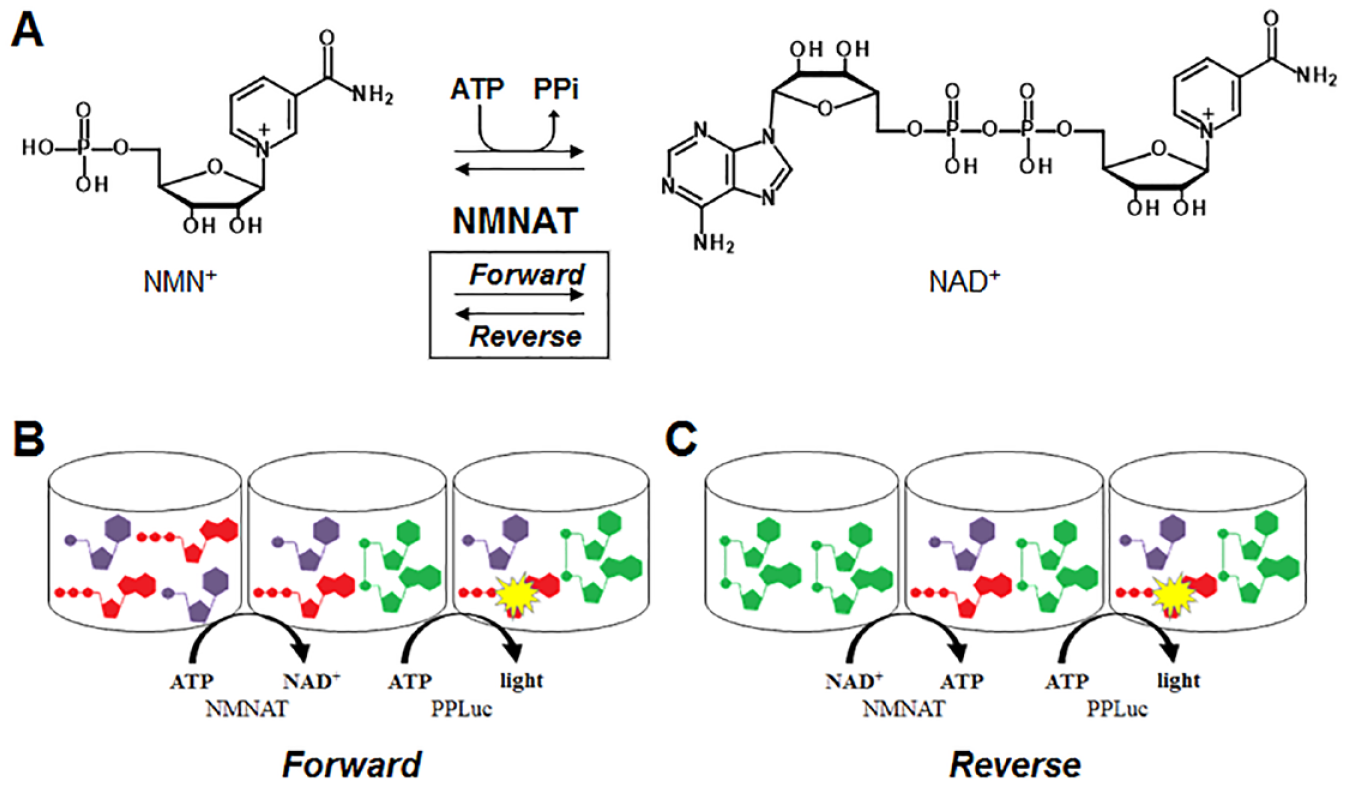
NMNAT1 reaction scheme and assay paradigm. (
The human genome encodes for three isoforms of NMNAT, termed NMNAT1, NMNAT2 and NMNAT3, which have distinct subcellular localizations: NMNAT1 in the nucleus, NMNAT2 in the cytosol and Golgi, and NMNAT3 in the cytosol and mitochondria.2,12 NMNAT has potential as a drug target for cancer if potency and selectivity for cancer cells can be achieved.13,14 NMNAT also has potential as a drug target for neurodegenerative diseases and aging if activators can be identified. Guarente states that “a model emerges in which aging is associated with PARP [poly (ADP-ribose) polymerase] activation, NAD+ depletion, sirtuin inactivation, mitochondrial dysfunction, and degeneration of cells and tissues, and where this calamity can be corrected or forestalled by supplementation with NAD+ precursors.” 15 Activation of NMNAT may provide an additional means to increase the concentrations of NAD formed from the precursors. Epigallocatechin gallate was discovered to activate NAD production by NMNATs, in an isoform-specific manner, nearly doubling NMNAT2 activity, while affecting NMNAT1 by 10%. 2 However, potent and selective inhibitors of NMNATs have yet to be described.
Our goal in this work was to develop an assay format and follow-up paradigm suitable to screen for new modulators of NMNAT and efficiently characterize the molecular mechanism of action (MMOA) for time dependence and competition with substrates. The assays reported in literature used to characterize NMNAT activity have identified compounds with modest activity against NMNAT.
2
These assays used low-throughput screens of substrate analogs and inhibitors chosen for activity against other NAD-related enzymes. The assay formats used for the forward reaction included discontinuous and continuous tandem reactions with alcohol dehydrogenase, high-performance liquid chromatography (HPLC),
16
or inorganic pyrophosphatase.
4
For the reverse reaction (NAD+ + PPi → NMN+ + ATP) (
As noted above, our goal also included developing a paradigm to enable early characterization of the MMOA of the inhibitors. Early understanding of the MMOA of modulators identified in target-based screens will facilitate optimization of molecules with the most suitable mechanistic properties. For example, it is important when starting with a target-based screen to differentiate molecules as competitive inhibitors, noncompetitive inhibitors, or activators, and whether they are time dependent or not. Establishing assays to identify compounds for time dependence also may identify potent compounds that may be missed and that have properties that could lead to increased selectivity. We have hypothesized that time-dependent inhibition in a nonequilibrium system can increase the selectivity of an inhibitor, due to kinetic selectivity, and thereby increase the therapeutic index and potential utility of a medicine.20–23
The goals of this work were achieved with the development a bioluminescent assay using Photinus pyralis ATP-dependent luciferase (PpLuc) and luciferin for NMNAT1 in a 384-well plate format that measures both the forward (
Materials and Methods
Reagents
The cyclic alkylaminoluciferin CycLuc1 and reduced coenzyme A (CoASH) were purchased from EMD Millipore (Darmstadt, Germany).
Human NMNAT1 and PpLuc were prepared by Biozilla as detailed below.
All other reagents, unless otherwise noted, were purchased from Sigma.
Expression and Purification of Recombinant NMNAT1
Full-length NMNAT1 (accession no. UniProtKB–Q9HAN9) was cloned into the pET21d expression vector in-frame with a C-terminal 6× His tag. BL21 (DE3) Escherichia coli were transformed with the construct and cultured in lysogeny broth (LB) to an optical density (OD; at 590 nm) of 0.9. Expression of NMNAT1 was induced with isopropyl-β-D-1-thiogalactopyranoside (0.5 mM) for 17 h at 24 °C. Cells were harvested and resuspended in lysis buffer (50 mM HEPES [pH 7.5], 300 mM NaCl, 10% glycerol, 20 mM imidazole, 10 µM phenylmethylsulfonyl fluoride, and 2 mM β-mercaptoethanol). The bacteria were lysed by sonication and freeze/thaw cycles. Insoluble protein was removed by centrifugation (12,000g, 30 min, 4 °C) and the soluble fraction loaded onto a nickel immobilized metal affinity chromatography (Ni-IMAC) column. The column was washed with 50 mM HEPES (pH 7.5), 300 mM NaCl, 10% glycerol, 40 mM imidazole, 0.02% Triton X-100, and 2 mM β-mercaptoethanol followed by 50 mM HEPES (pH 7.5), 300 mM NaCl, 10% glycerol, 80 mM imidazole, and 2 mM β-mercaptoethanol. Recombinant NMNAT1 was eluted from the IMAC column with 4.5 mL of 25 mM HEPES (pH 7.5), 300 mM NaCl, 10% glycerol, 500 mM imidazole, and 2 mM β-mercaptoethanol. Eluted NMNAT1 was further fractionated by size-exclusion chromatography (SEC) using a Superdex-200, 10/300 GL column preequilibrated with SEC running buffer, 50 mM Tris-HCl (pH 7.5), and 100 mM NaCl. Fractions were collected, and a portion of each was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) to identify the fractions containing NMNAT1. A single band corresponding to the molecular weight of NMNAT1 was eluted in fractions 13–15 and confirmed to be active NMNAT enzyme by activity measurement (
Expression and Purification of Recombinant PpLuc
Full-length PpLuc (accession no. UniProtKB–Q27758) was cloned into the pET21d expression vector in-frame fused to myelin basic protein to its N-terminal side and to a 6× His tag to its a C-terminal side. BL21 (DE3) E. coli were transformed with the construct and cultured in LB to an OD (at 590 nm) of 0.9. PpLuc was purified through a Ni-IMAC as described above for NMNAT. The eluate from the nickel column contained one major band on SDS-PAGE and was dialyzed against 20 mM Tris-Cl (pH 8.0), 20% glycerol, and 1 mM DTT, and used without further purification (
NMNAT1 Enzyme Assay
Preparation of Detection Reagent Mixture
The ATP detection reagent was prepared by mixing PpLuc (26 µg/mL),
NMNAT1 Assay
Unless otherwise noted, in a final volume of 20 µL the reaction consisted of either 6–10 nM (forward reaction) or 0.5–1 nM (reverse reaction) NMNAT1 and an appropriate substrate in NMNAT buffer (15 mM HEPES adjusted to pH 7.4, 20 mM NaCl, 10 mM MgCl2, 1 mM EGTA, and 0.02% [v/]) Tween-20). The substrates used for the forward reaction were ATP (20 µM) and NMN+ (100 µM), and substrates used for the reverse reaction were NAD+ (100 µM) and pyrophosphate (100 µM). The reaction was allowed to proceed at ambient room temperature (about 23 °C) and was stopped by the addition of 45 µL of the detection reagent. Following 20 min of additional incubation, the luminescent signal coming from the amount of ATP either remaining (forward reaction) or generated (reverse reaction) was measured using a Luminoskan Ascent plate reader (Thermo Labsystems, Waltham, MA).
High-Throughput Screening
HTS of NMNAT1 inhibition was performed in the forward direction (NMN+ → NAD+), by measuring depletion of ATP-proportional light. Inhibitors (5 µL of 80 µM 8% DMSO [v/v]) and NMNAT1 (5 µL of 40 nM) were added to 384-well plates followed by substrates (10 µL of 40 µM ATP and 200 µM NMN+). All liquid transfers were made using a Biomek 2000 automated liquid handling system (Beckman Coulter, Indianapolis, IN). The final concentrations of the reagents were 10 nM NMNAT1, 20 µM ATP, 100 µM NMN+, 20 µM inhibitor, and 2% (v/v) DMSO. Each plate contained multiple wells for control and ATP standards that had 2% DMSO and no inhibitor. All compounds were tested in duplicate. The reaction was initiated by the addition of the substrate and was allowed to proceed for 55 min at ambient room temperature. The reaction was terminated by adding 45 µL/well of the detection reagent, and the resulting luminescence was measured after 20 min of additional incubation using a Luminoskan Ascent plate reader. The NMNAT-catalyzed metabolism was calculated by eq 1A:
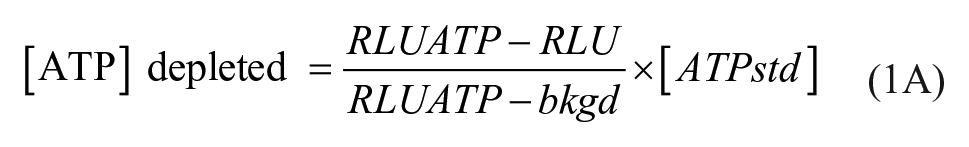
where RLU and RLUATP are relative light signals for the sample and ATP standard, respectively, bkgd is a background light signal of 100 µM NMN+ and luciferase reagent in the absence of NMNAT1 and ATP, and [ATPstd] is the concentration of ATP used with no NMNAT1.
Follow-Up Screen
Follow-up assays were run exactly as described above, but in addition to the forward reaction, inhibition of the reverse reaction (NAD+ → ATP) by compounds was also measured using 0.5 nM NMNAT, 100 µM NAD+, and 100 µM PPi as substrates, and the NMNAT-catalyzed ATP generation was calculated by eq 1B:
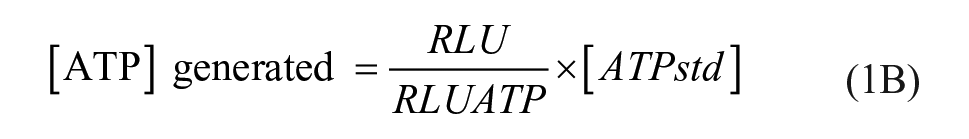
where RLU and RLUATP are relative light signals for the sample and ATP standard, respectively, and [ATPstd] is the concentration of ATP used with no enzyme.
Counterscreen
Compounds selected from HTS for follow-up were tested for their effect on the PpLuc assay in the absence of the NMNAT1. NMN+ or NAD+/PPi were included in background and normalization data points to account for any loss of signal due to these entities in the luciferase reaction mixture.
None of the active compounds selected for follow-up inhibited PpLuc activity. 2,3-Dibromo-1,4-naphthoquinone (DBNQ) was also tested for degradation of ATP upon preincubation; without enzyme, no change in ATP quantitation was noted relative to ATP signals without preincubation with DBNQ.
Molecular Mechanism of Action Assays
Time Dependence
A time dependence screen was performed with selected inhibitors from the primary screen (HTS). Time-dependent studies were performed by preincubating 2 and 20 µM test compounds with NMNAT1 for 45 min at ambient room temperature using the reaction parameters described above, NMNAT1-catalyzed reactions were initiated with the addition of the substrate pair (100 µM NMN+ and 20 µM ATP) in 20% volume (4 µL), and the reaction was allowed to proceed for an additional 10 min. Time dependence was noted by an increase of inhibition at the 20 µM inhibitor with preincubation and a dose response between 2 and 20 µM compound concentrations.
Dose–response studies for time dependence were conducted with dihalo-naphthoquinones (DHNQs) by generating IC50 plots. Following 45 min of preincubation, NMNAT1-catalyzed forward and reverse reactions were initiated and measured as described above.
DBNQ association experiments were conducted by varying the preincubation time of DBNQ and NMNAT1 from 0 to 45 min with 1 µM DBNQ and 6 nM NMNAT1 (forward reaction) and 1 nM NMNAT1 (reverse reaction), followed by the addition of substrate pairs and 10 min of incubation.
Evaluation of Mechanism of Action
Inhibition can be competitive, noncompetitive, or uncompetitive in relationship to substrates. The mechanism of action was evaluated by preincubating with and without substrates. The activity of competitive inhibitors will decrease in the presence of competing substrate concentrations, will be unchanged for noncompetitive inhibition, and will increase with uncompetitive inhibition. To evaluate the mechanism of action of the inhibitors, test compounds at IC50 and NMNAT were preincubated for 45 min without and with substrates (ATP and NMN) and then enzymatic activity was measured. A loss of NMNAT1 inhibitory activity upon co-preincubation with one substrate was interpreted as competitive behavior of the inhibitor with respect to that substrate.
Data Analysis
For dose and time dependence experiments, Michaelis–Menten plots, IC50 values, and association experiments, data were fit to respective graphs using GraphPad Prism 6 (La Jolla, CA). Statistical values were derived from the BEST test. 25 Results with p < 0.05 were considered statistically significant. HTS data and corresponding histograms were analyzed using built-in software from Collaborative Drug Discovery (CDD; Burlingame, CA).
Results
Development of Bioluminescent NMNAT1 Assay
Discontinuous enzymatic assays involving a coupled ATP-dependent luciferase to measure light-proportional ATP have been successful for kinases and other classes of enzymes.
26
We adapted a tandem PpLuc step to quantitate ATP in the reaction mixture. Using sulfate and CoASH to slow the light signal decay of PpLuc,27,28 we stabilized light signals to achieve linearity of signal and optimized to it our chosen ATP concentration of 20 µM, approximating 0.5 Km. Standard curves were generated to simulate NMNAT-catalyzed metabolism of ATP, including NMN+ at assay concentrations, and NAD+ and PPi concentrations consistent with consumption of ATP and NMN+ during the enzymatic assay (
Activity of NMNAT1
Human NMNAT1 was expressed in E. coli with an N-terminal His tag and purified to homogeneity as described in Materials and Methods. A discontinuous in vitro bioluminescent assay was used to quantify ATP from light generated by PpLuc.
26
The assay was optimized to achieve linearity of activity with respect to NMNAT1 concentration and time in the forward (
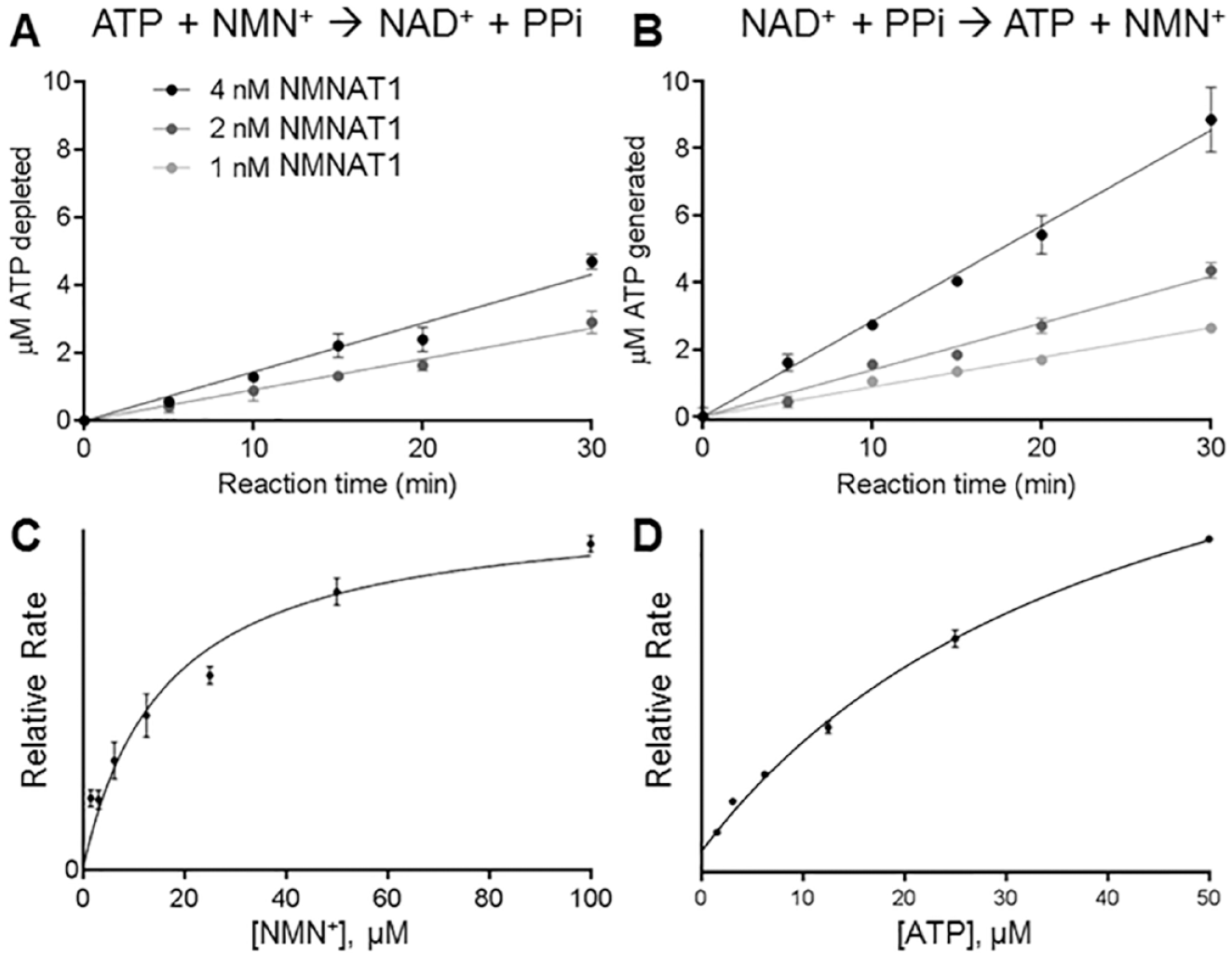
Enzyme kinetics of NMNAT1. Linearity of NMNAT1-catalyzed reaction with respect to time and protein concentration for (
HTS of Oncology Libraries with NMNAT1
The conditions for screening compounds in the 384-well format were chosen to ensure linearity with time and protein as well as provide a sufficient signal-to-noise ratio. The compounds were tested at a single concentration (20 µM) in duplicate.
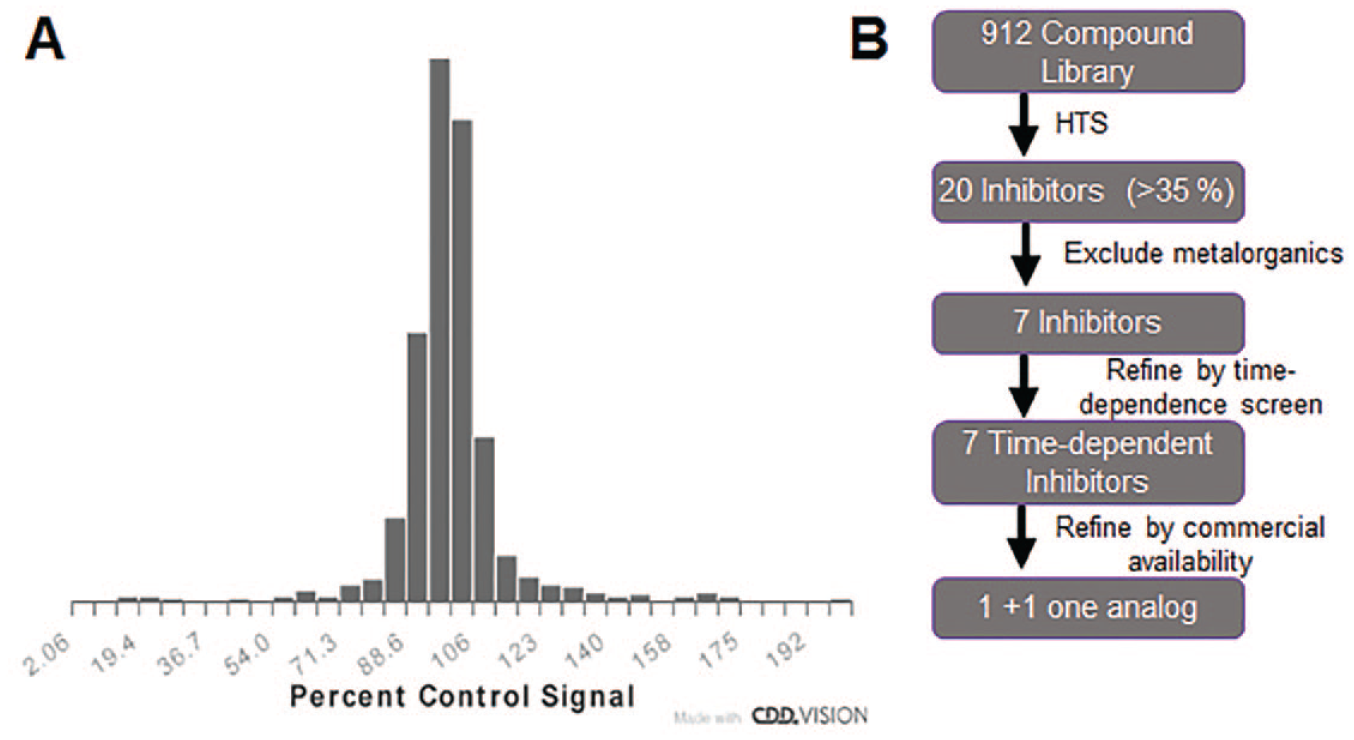
NMNAT1 inhibitor screen paradigm and results. (
Time-Dependent Assay Follow-Up on HTS Actives
Of the 20 hits from the HTS, 13 were metalorganic compounds and were excluded from follow-up. To access time-dependent inhibition of NMNAT1 by the remaining seven molecules, a secondary screen that included a 45 min preincubation with 20 and 2 µM compounds was performed. From this screen, all of these compounds were identified as time-dependent inhibitors by the increased inhibition following preincubation (
Activity of HTS Hits and Analogs without and with Preincubation.
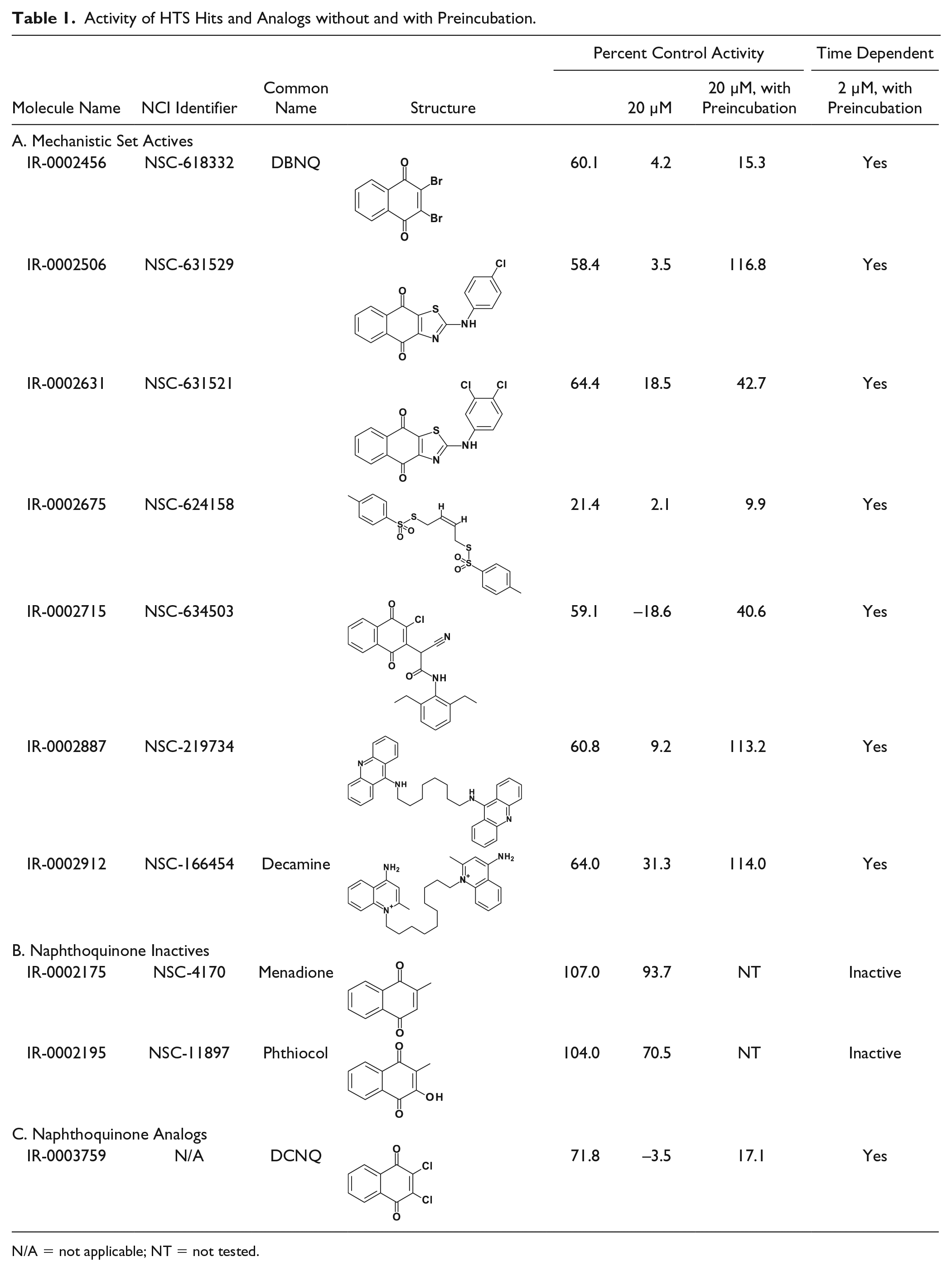
N/A = not applicable; NT = not tested.
IC50 Values of the DHNQs without and with Preincubation
The IC50 values were determined in reactions without and with preincubation and initiated by the addition of substrates NMN+ and ATP for the forward reaction and NAD and PPi for the reverse reaction. Following preincubation with NMNAT, DBNQ and DCNQ had IC50 values of 0.76 and 1.17 µM, respectively, while their IC50 values were >20 µM without preincubation (
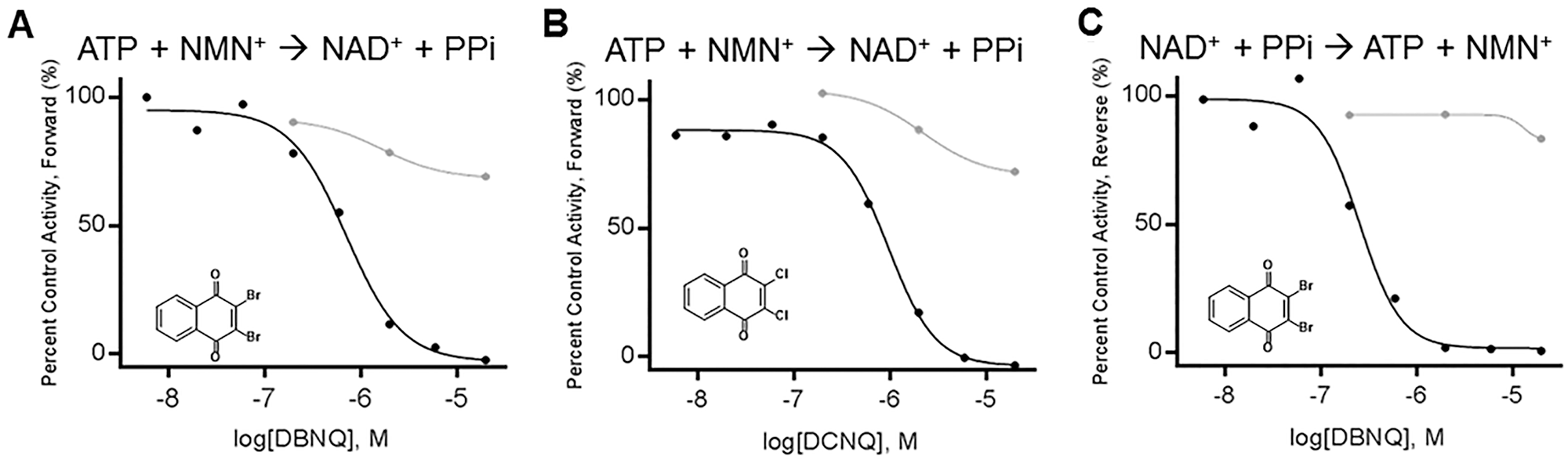
DHNQ dose–response on NMNAT1-catalzyed metabolism without (gray) and with (black) a 45 min preincubation prior to addition of substrate pair. (
Substrate Competition by DBNQ
Enzyme activity of DBNQ-preincubated NMNAT1 was partially recovered when co-preincubated with each NMN+ (p = 0.0536) and ATP, at 5 and 0.5 Km, respectively, indicating the competitive behavior of DBNQ with both substrates (
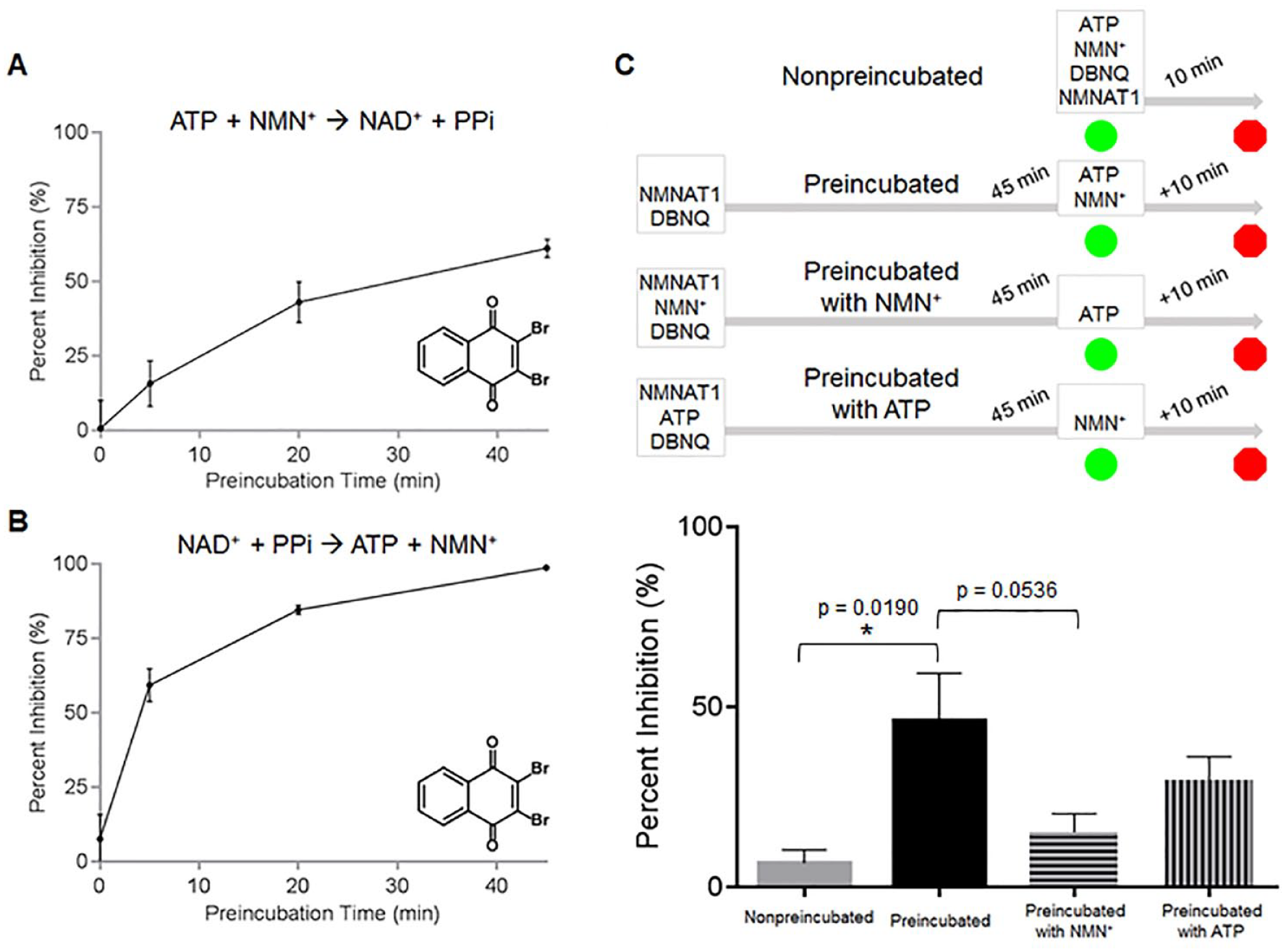
Mechanistic studies with NMNAT inhibitors. Time dependence of DBNQ. DBNQ (1 µM) and NMNAT1 were preincubated for indicated time periods and the reaction was allowed to proceed for 10 min following the addition of substrates (
Discussion
NAD biosynthesis is an important contributor to cellular homeostasis. It is hypothesized that selective inhibition is a potential target for cancers and increased biosynthesis for neurodegenerative diseases. Targeting the energetics of the cell, including NAD+ and ATP, has been hypothesized as therapy for cancer since the 1980s. 29
To our knowledge, a plate-based HTS assay method for screening NMNAT has not previously been reported. NMNAT inhibitors described from other laboratories require high concentrations to effect NMNAT activity. Gallotannin, the strongest inhibitor of NMNAT1 reported before this study, has an IC50 of 10 µM for NMNAT1 and shows slight selectivity for NMNAT3, with an IC50 of 2 µM.
2
Product analogs with polyphosphate insertions to NAD+ demonstrate mediocre inhibition toward NMNATs.
2
Most recently, a ureyl NMN+ analog, Vacor adenine dinucleotide, was found to have IC50 values for NMNAT2 and NMNAT3 of 20 and 463 µM, respectively, with no detectible effect on NMNAT1 activity.
30
Not surprisingly, our screen of the NCI mechanistic set, which contains several adenosine analogs and other ATP-competitive inhibitors for kinases, reflected this poor inhibition by substrate analogs with little activity against NMNAT1 at 20 µM (
The screening method also included a mechanistic follow-up. The inhibitors of NMNAT were evaluated for time dependence by preincubation of enzyme with inhibitor and evaluating the effect of preincubation on activity (
As part of our ongoing efforts to identify new treatment molecules for rare diseases, including cancers, we have been investigating approaches to identify new MMOAs early in the discovery process. Much of our appreciation for the importance of MMOA to a therapeutic index is anecdotally based on the observations that many approved drugs have MMOAs that allow for efficient coupling, including slow dissociation kinetics and irreversible binding. 22 Transitioning from reversible equilibrium competition binding to nonequilibrium competition binding will lead to noncompetitive behavior that will lower the concentration of drug required for efficacy in a system with high levels of competing substrate. The significance of this is that the lower drug concentrations will lead to better selectivity and an increased therapeutic index. Early recognition of mechanistic differentiation of new drugs would be useful to inform optimization and development. 31 In this report, we demonstrate that the methods to mechanistically evaluate compounds are straightforward and can be readily incorporated into a follow-up paradigm.
In summary, we report a bioluminescent assay to measure the activity of NMNAT. This versatile assay is useful in plate format and is suitable for screening for new modulators of this enzyme. In screening a small library, time-dependent inhibitors that are competitive with NMN+ and ATP were identified. These are the most potent NMNAT1 inhibitors reported to date.
Supplemental Material
Supplemental_Material_for_NMNAT_Screening_Assay__by_Haubrich_et_al – Supplemental material for Development of a Bioluminescent High-Throughput Screening Assay for Nicotinamide Mononucleotide Adenylyltransferase (NMNAT)
Supplemental material, Supplemental_Material_for_NMNAT_Screening_Assay__by_Haubrich_et_al for Development of a Bioluminescent High-Throughput Screening Assay for Nicotinamide Mononucleotide Adenylyltransferase (NMNAT) by Brad A. Haubrich, Chakk Ramesha and David C. Swinney in SLAS Discovery
Footnotes
Acknowledgements
We thank the National Cancer Institute for compound libraries. The authors wish to acknowledge Biozilla led by Dr. David Chereau for providing NMNAT1 and PpLuc. The authors also wish to acknowledge Dr. Shuangluo Xia for helping to conceptualize the NMNAT assay; Dr. Brian S. Blais, Bryant University, for assistance with statistical analyses; and Dr. Peter Gund, CDD, for critical reading of the manuscript.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a grant from NIAID RO1AI103476.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.