
Abstract
Mass spectrometry-based phosphoproteomics holds promise for advancing drug treatment and disease diagnosis; however, its clinical translation has thus far been limited. This is in part due to an unstandardized and segmented sample preparation process that involves cell lysis, protein digestion, peptide desalting, and phosphopeptide enrichment. Automating this entire sample preparation process will be key in facilitating standardization and clinical translation of phosphoproteomics. While peptide desalting and phosphopeptide enrichment steps have been individually automated, integrating these two extractions and, further, the entire process requires more advanced robotic platforms as well as automation-friendly extraction tools. Here we describe a fully automated peptide desalting and phosphopeptide enrichment method using IMCStips on a Hamilton STAR. Using our established automated method, we identified more than 10,000 phosphopeptides from 200 µg of HCT116 cell lysate without fractionation with >85% phosphopeptide specificities. Compared with titania-based Spin Tip products, the automated IMCStips-based method gave 50% higher phosphopeptide identifications. The method reproducibility was further assessed using multiple reaction monitoring (MRM) to show >50% phosphopeptide recoveries after the automated phosphopeptide extraction with coefficients of variation (CVs) of <20% over a 3-week period. The established automated method is a step toward standardization of the sample preparation of phosphopeptide samples and could be further expanded upon to create a fully automated “cells to phosphopeptides” method.
Introduction
Protein phosphorylation is an important posttranslational modification used to communicate vital signals throughout the cell.1–5 Kinases and phosphatases are used within the cell to turn “off” or “on” signaling cascades, leading to a variety of critical intracellular events, such as cell proliferation and apoptosis.3,4,6 Understanding the roles that protein kinases and phosphatases play by mapping protein phosphorylation is of particular interest, as many disease states, such as cancer and neurodegenerative diseases, stem from aberrant signaling events. 7 Such interest is exemplified by the number of kinase inhibitors approved by the Food and Drug Administration (FDA) for treating cancers and other diseases. 8 With the advent of modern mass spectrometry (MS), researchers are now provided with more complex phosphorylation patterns from thousands of proteins in each mass spectrometric analysis.9–11 Further developments in labeling techniques, such as stable isotope labeling with amino acids in cell culture (SILAC) and Tandem Mass Tag (TMT) labeling, have allowed researchers to reliably quantify changes to these proteins in response to stress, treatments, or novel compounds.12–16 Recent work isolating phosphoproteins in extracellular vesicles suggests that phosphorylated proteins are potential biomarkers for early breast cancer detection. 17
While MS-based phosphoproteomics has advanced over the past 10 years, the field is still plagued by a labor-intensive sample preparation process, limiting its translational impact. Due to the low abundance of phosphopeptides in biological samples, phosphopeptide analysis requires a multistep sample preparation process prior to mass spectrometric analysis, starting from cell lysis, protein digestion, peptide desalting, and phosphopeptide enrichment. Each step can introduce variations that can obfuscate mass spectrometric results. The challenge to provide a consistent and automated workflow needs to be addressed before this powerful analytical process can be further expanded and possibly translated to more routine clinical and pharmaceutical applications.
Both immunoaffinity and IMAC resin-based phosphopeptide enrichment methods have been reported using modified magnetic beads or resins packed in pipette tips to be used on automated liquid handling systems (ALHSs).18–22 A main limitation of these approaches is that they use ALHSs that are designed for single-function operations, such as peptide desalting or phosphopeptide enrichment, making automating multiple extraction processes difficult, and thus segmenting the sample preparation process. 23 To address this limitation in this study, the Microlab STAR from Hamilton Robotics was selected as it is able to automate multiple extraction processes in a single automation method. While magnetic beads can be integrated onto the STAR system, the method requires a robotic arm and magnetic stand to be incorporated into the system. Spin column-based chromatography products require coupling with a third-party software and a centrifuge. Adding these additional accessories would increase capital costs and reduce the overall workspace on the robotic platform. Hence, we selected IMCStips, which incorporate various chemistries in pipette tips to perform extractions without needing to further upgrade the automation platform.
Pipette tip-based extractions have been popularized over the last two decades, mostly involving applications ranging from food to forensic analysis and, more recently, protein purification. 24 Most SPE-packed pipette tips are tightly restricted by two filters, which results in higher back pressures, and they are prone to clogging. One popular tip-based SPE is a monolith-based pipette tip that is commonly used for desalting peptides prior to spotting with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). 25 In comparison with the other pipette extractions, IMCStips perform dispersive solid-phase extraction (dSPE) in a pipette tip with bidirectional flow for a faster workflow. This difference has been shown to lead to lower back pressure and faster workflow, making dSPE automation friendly. 24
Here, we report the first integrated workflow to couple desalting and phosphopeptide enrichment by using this novel dSPE in a pipette using a single program on an automated liquid handling platform to provide greater consistency for complex workflows (
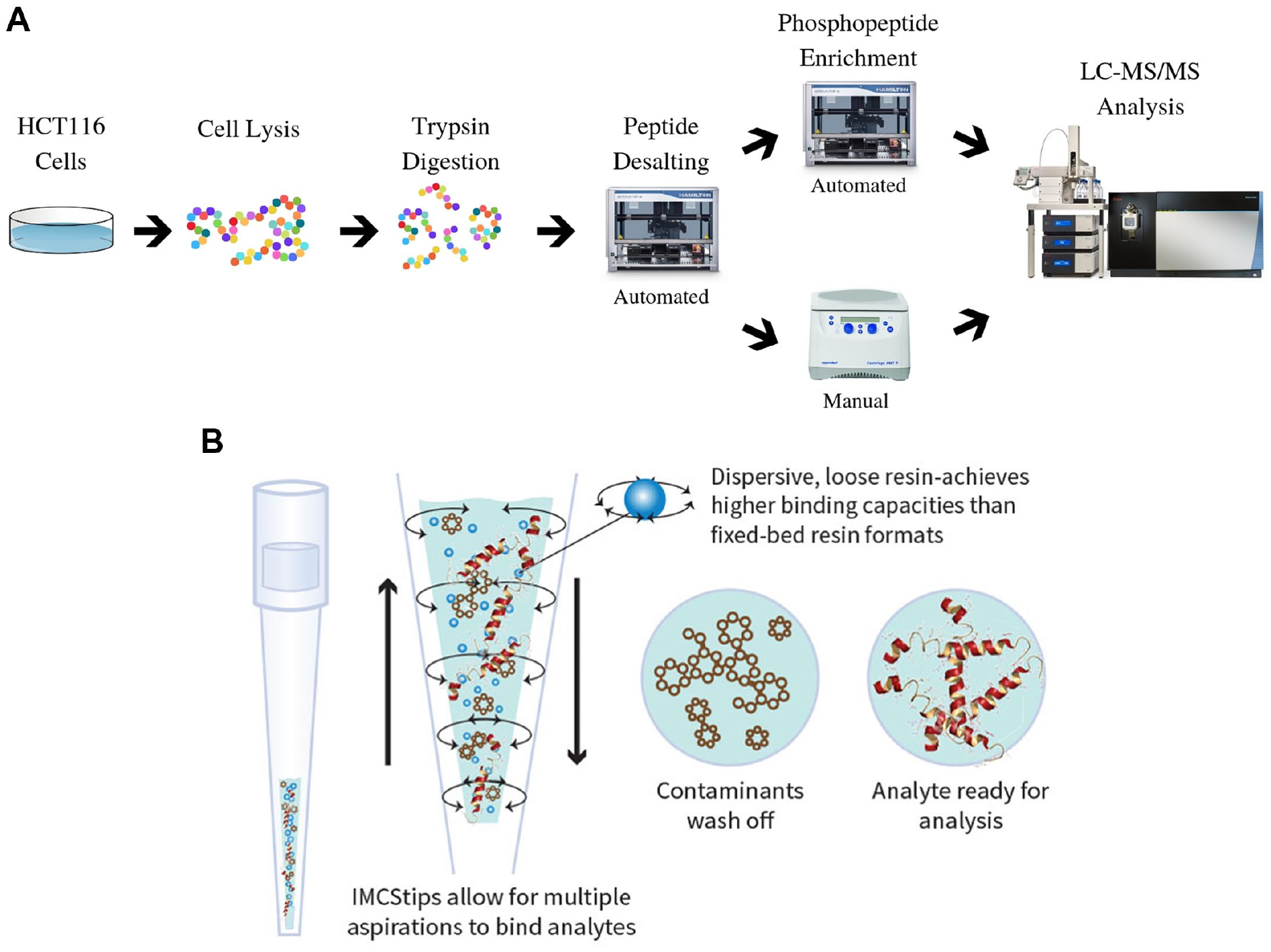
(
Materials and Methods
Cell Culture and Treatment
HCT116 cells were grown in Gibco DMEM/F-12, containing 15 mM HEPES and supplemented with 10% fetal bovine serum (FBS). Cells were passaged by trypsin/EDTA treatment at 80%–90% confluence. The cells were passaged at least five times prior to a treatment with 10 mM H2O2/Na3VO4 for 30 min. This cell treatment is known to activate several different phosphorylation signaling cascades and protect the phosphorylation sites from active phosphatases, making the treatment ideal for establishing the phosphopeptide enrichment method.26,27 After treatment, the cells were washed three times with cold 1× phosphate-buffered saline (PBS). The cells were collected by scraping from the dish, and pelleted by centrifuging for 5 min at 500g. The supernatant was removed and the cell pellets were stored at –80 °C until further processing.
Cell Lysis
For HCT116 cell lysis, ~1 × 108 cells were lysed with 1 mL of RIPA buffer (Thermo Scientific Pierce, Rockford, IL) containing 1× Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific Pierce). Proteins were reduced with the addition of 10 mM TCEP at 56 °C for 30 min, and then alkylated with the addition of 25 mM iodoacetamide for 30 min in the dark at room temperature (20–25 °C). Next, 1.5 mL of ice-cold acetone was added to the sample to precipitate the proteins. The mixture was placed at –80 °C for 30 min, followed by –20 °C for 30 min. The sample was then centrifuged for 5 min at 20,000g and the supernatant was discarded. The protein pellet was washed using 1 mL of ice-cold acetone and centrifuged for 5 min at 20,000g. The supernatant was then discarded. The resulting pellet was suspended in 50 mM ammonium bicarbonate to a final concentration of 5 mg/mL, followed by overnight trypsin digestion (1:50 enzyme to protein by mass ratio) at 37 °C using MS Grade Trypsin Protease (Thermo Scientific Pierce). The sample was then acidified to 1% trifluoroacetic acid (TFA) and then centrifuged for 5 min at 5000g. The supernatant was then aliquoted and stored at –80 °C until required for phosphopeptide enrichment.
Phosphopeptide Enrichment of HCT116 Protein Digests
Protein digest was thawed on ice for 30 min. Aliquots of 20 µL of protein digest were then diluted with 1% TFA to a final volume of 200 µL and transferred to a 1 mL 96-well plate. The automated phosphopeptide enrichment used 300 µL of IMCStips (IMCS, Irmo, SC), which contain loosely packed resins that are specific to the workflow. All reagents were poured into large reagent troughs, and the program was designed to aliquot solutions and buffers into specific positions used for the enrichment process. The peptide desalting portion of the extraction was performed using pipette tips containing 5 mg of reverse-phase (RP) resin (polystyrene crosslinked with divinylbenzene). The desalting resin quantity and liquid handling program were screened to ensure that more than 90% recovery was achieved (
Additional details of the automated extraction process, such as time associated with each step, the amount of solution aspirated and dispensed, the cycle number (number of times the solution is aspirated and dispensed) for each step, the aspiration and dispense speeds for each step, and the buffers used for each step, are specified
Database Search and Analysis
For the initial optimization of the phosphopeptide enrichment method, an LTQ-Orbitrap Velos Pro (Thermo Fisher) was used for MS analysis. Once the method was optimized, Orbitrap Fusion (Thermo Fisher) was used for MS analysis. The MS methods for each instrument can be found in the supplemental information. The raw data files obtained from LTQ-Orbitrap Velos Pro and Orbitrap Fusion were searched against a human database using SEQUEST on Proteome Discoverer (Version 2.2, Thermo Fisher). The peptide precursor mass tolerance was set to 10 ppm, and the fragment mass tolerance was set to 0.6 Da. Search criteria included a static modification of cysteine residues of +57.0214 Da and a variable modification of +15.995 Da to include potential oxidation of methionine and a modification of +79.966 Da on serine, threonine, or tyrosine for the identification of phosphorylation (three modifications allowed per peptide). Searches were performed with full tryptic digestion and allowed a maximum of two missed cleavages on the peptides analyzed from the sequence database. False discovery rates (FDRs) were set to less than 1% for each analysis at the peptide-spectrum match (PSM) level. Phosphorylation site localization from collision-induced decomposition (CID) spectra was determined by PhosphoRS on Proteome Discoverer 2.2. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012583. All Venn diagrams for peptide overlap were generated using Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/). Functional protein association networks were analyzed using String software (https://string-db.org/). The confidence view was assigned a score of 0.4, indicating medium confidence. The GRAVY values of phosphopeptides were calculated using the GRAVY calculator (http://gravy-calculator.de/index.php) and the pI values were calculated using the Compute pI/Mw tool (https://web.expasy.org/compute_pi/).
Phosphopeptide Enrichment for MRM Assay
Four purified phosphopeptides from Anaspec (Fremont, CA) and two purified phosphopeptides from New England Peptide (Gardner, MA) were screened for compatibility with the multiple reaction monitoring (MRM) assay based on stability and ionization efficiencies. Three phosphopeptides were selected as optimal candidates for monitoring by MS/MS. Four additional nonphosphopeptides were added to the assay to determine recovery of nonphosphorylated peptides. The standard sample comprised 2 pmol of 3 phosphorylated and 2 pmol of 4 nonphosphorylated peptides fortified with 50 µg of trypsin-digested bovine serum albumin (BSA). The mass spectrometric parameters for the seven peptides are provided in
Multiple Reaction Monitoring
The MRM assay was used to analyze phosphopeptide samples after the automated enrichment. For the MRM assay, a TSQ Endura (Thermo Fisher) coupled to a Vanquish UHPLC was used. RP separation was performed using a Waters (Milford, MA) BEH C18 analytical column (100 × 2.1 mm, 1.7 µm) heated at 40 °C with a liquid chromatography (LC) gradient of 2%–35% B (0.1% formic acid in acetonitrile) for 10 min. TSQ Endura was run under optimized conditions with a Q1 and Q3 resolution of 1.2 and 0.7 FWHM (full width at half maximum), respectively, and CID gas was set to 1.5 mTorr. All raw files were processed using Skyline 4.0. All samples were compared and quantified based on a neat sample injected before and after each sample set.
Results
Optimization of Phosphopeptide Enrichment
The IMCStips-based automated method follows a conventional “bind, wash, elute” workflow in which different solutions are aspirated and dispensed. Each step in the extraction method requires optimization in terms of buffers used, volume of buffer used, incubation time, and aspiration/dispense speed. For the sample binding portion, it was necessary to optimize the resin-to-peptide amount ratio. A fixed quantity of resin was used with varying amounts of cell lysate and a 1:20 ratio for the PolyTi and a 1:40 ratio for the ZrO2 were selected (
Phosphopeptide Enrichment of HCT116 Protein Digests
Each sample aliquot for analysis by Orbitrap Fusion comprised 200 µg of peptide from HCT116 protein digests. The cell culture, lysis, protein precipitation, and digests were all performed identically in bulk prior to splitting the samples into multiple aliquots for comparison of Spin Tips and IMCStips. For PolyTi and ZrO2 resins in IMCStips, the average phosphopeptide identifications were 10,191 and 9796, respectively (
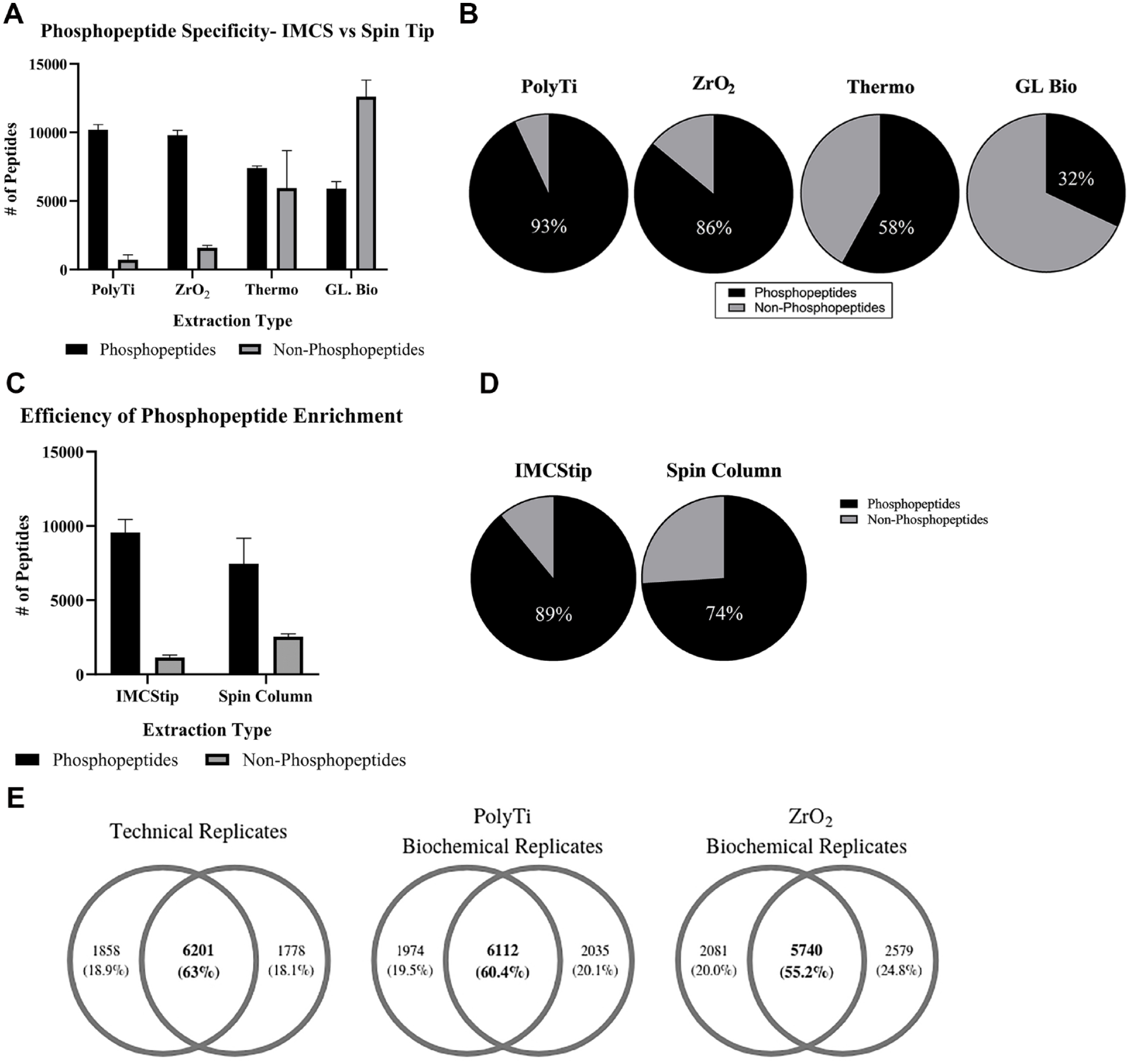
(
The reproducibility of phosphopeptide enrichment methods was assessed using two biochemical replicates. Technical reproducibility was obtained by injecting a single sample that was divided in half and injected sequentially. The peptide overlap for the technical replicate from this “split” sample was 63% (
Given the intrinsic limitation of DDA, the reproducibility of the established automated phosphopeptide enrichment was further assessed using an in-house MRM assay with three phosphopeptides and three nonphosphopeptides. For this initial work, several commercially available phosphopeptides were screened, but these three phosphorylated peptides provided sufficient MS signal intensities. For three phosphopeptides, PKC substrate 4 from Anaspec, MAPK1 from New England Peptide, and MAPK14 from New England Peptide, the signal intensities were unreliable and were therefore not used in the assay. The sequence of the seven peptides that were used for the MRM assay are listed in
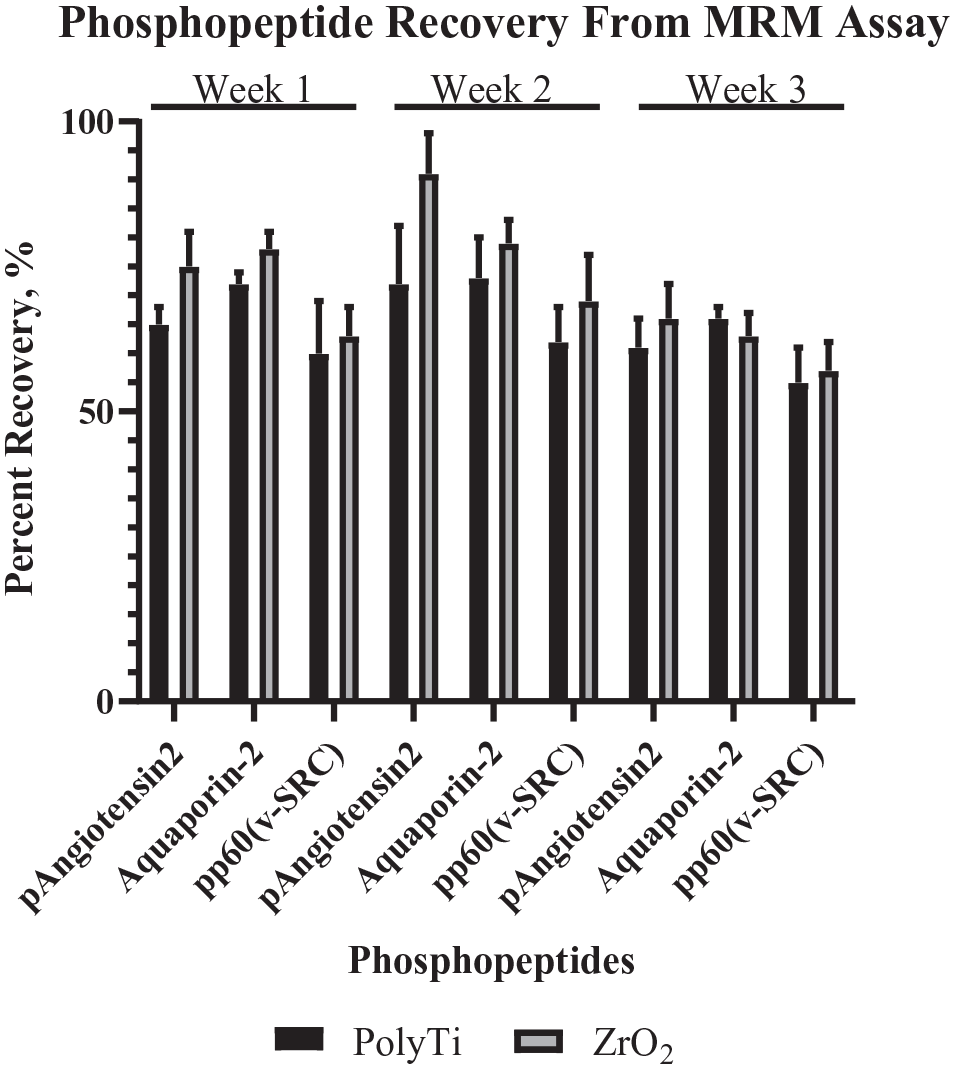
The average recovery for each phosphopeptide from three different extractions performed once a week over 3 week using two different resins (n = 8 per resin type; data are mean ± SD).
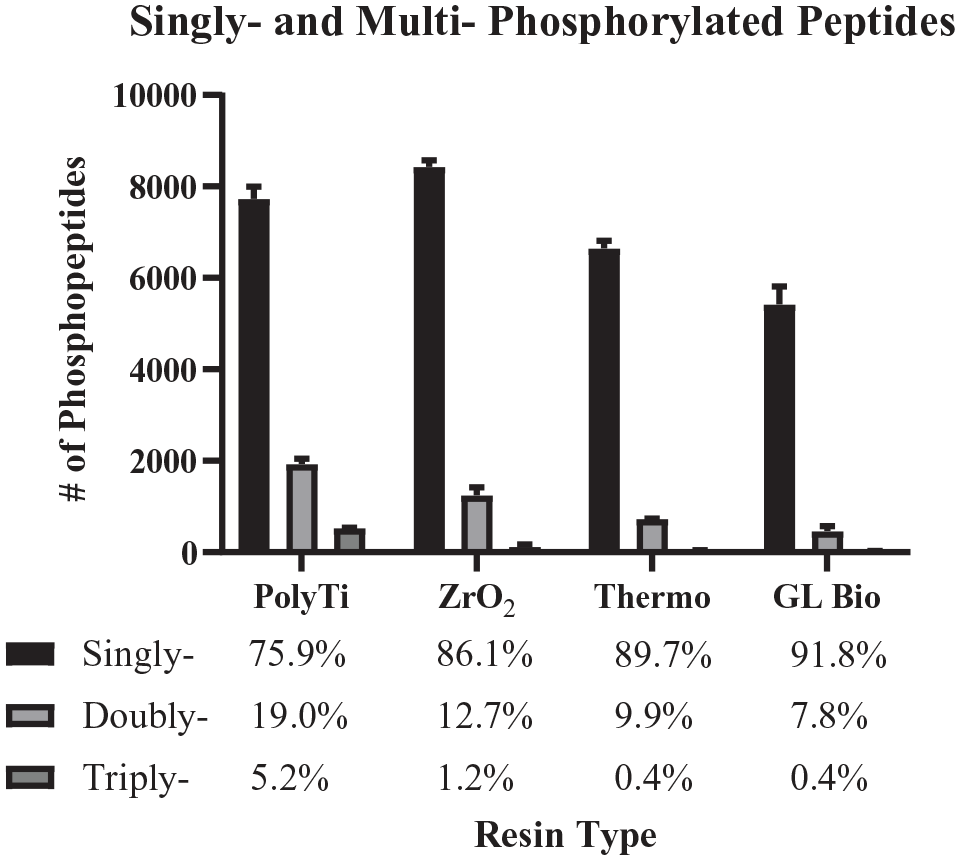
The distribution of singly, doubly, and triply phosphorylated peptides identified from 200 µg of HCT116 protein digest from the different extraction types (n = 2 per extraction type; data are mean ± SD).
Comparison of Resins
Zirconium, titanium, and iron are common metals used for phosphopeptide enrichment. Since zirconium- and titanium-based chemistries were available for dSPE in tips; the differences between the two resins using the same automated workflow were further investigated. The pool of singly, doubly, and triply phosphorylated peptides from the two different resins was minor, where 86% of phosphopeptides identified with the ZrO2 resin were singly phosphorylated compared with 76% for PolyTi resin. For PolyTi resin, on average, 24% of the phosphorylated peptides identified were multiply phosphorylated compared with 14% for ZrO2 and less than 10% for both TiO2-based Spin Tips (
The peptide overlap between PolyTi and ZrO2 (pooled peptide identifications from two biological replicates for both resin types) showed a slight decrease in peptide overlap compared with the biological replicates (
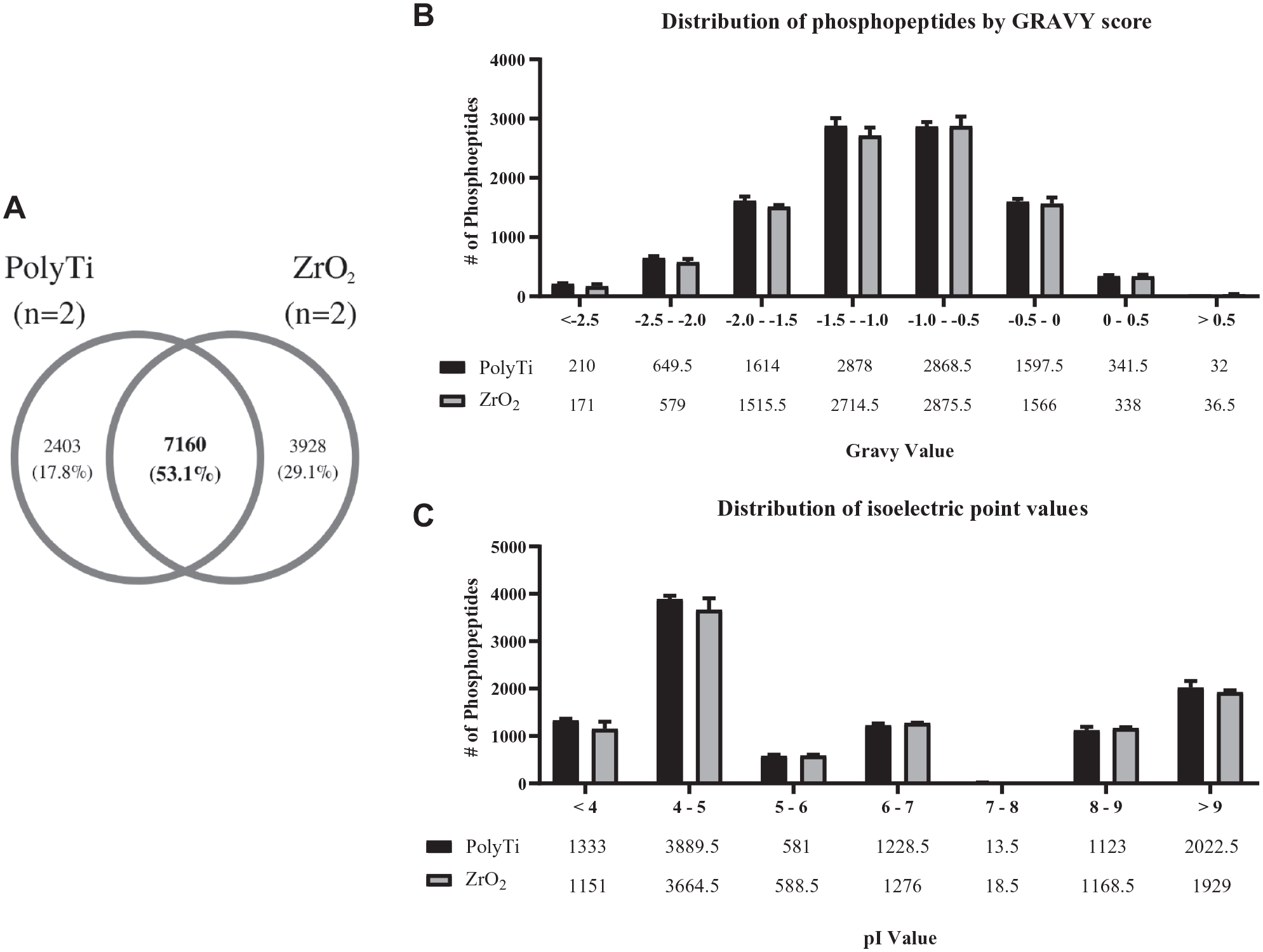
(
Discussion
As part of the standardization process, the entire workflow was unified with a single cell type of the human colon cancer cell line (HCT116). The selection of the colon cancer cell line as the initial starting point is due to the relatively well-established characterization of the cell line and the need for more effective colorectal cancer screens. 31 A recent report highlighted that higher incidence rates of colon cancer for adults age 20–39 years than for adults age 40–54 years raises concerns for the well-being of the general public. 32 Current novel biomarker research focuses on genetic, epigenetic, and protein markers as new screening modalities for early detection. This study is envisioned as an additional toolkit to expand that search for biomarkers, especially focusing on aberrant posttranslational modifications involving phosphorylation events.
To automate and optimize a nonsegmented desalting extraction and phosphopeptide enrichment method, we selected an IMCStips-based approach automated on the Hamilton STAR. The IMCStips did not require any additional hardware for the automation system and were easily programmed to perform the sample extractions based on a simple “bind, wash, elute” workflow. The Hamilton STAR is an ALHS that was compatible with the IMCStips and could easily accommodate the space needed to automate both the peptide desalting extraction and phosphopeptide enrichment in a 96-well format in one method. Further, a heat block can be added to the STAR system, which will be used in future work to automate the entire sample preparation process. The main advantage of the automated method is that only 10 min of hands-on time to load tips and reagents is required to set up the extraction. In contrast, the conventional Spin Tip-based method requires intermittent interventions throughout the entire extraction process. Additionally, the spin column-based method requires increased time with increased sample numbers due to manual loading of samples in the centrifuge and hand pipetting solutions for each sample. The IMCStips-based method requires the same amount of time for 1–96 samples. As mentioned above, the ideal sample preparation method would be a fully automated, “cells to phosphopeptides” workflow. The method presented here is able to automate the desalting and phosphopeptide enrichment portion of the sample preparation process; however, the current cell lysis and protein digestion protocol is not automation friendly as it requires several centrifugation steps. Recently, Humphrey et al. reported a cell lysis and protein digestion protocol that eliminates the acetone crash and subsequent protein pelleting, making the workflow more automation friendly. 33 This cell lysis and protein digestion protocol could be combined with the automated workflow presented here to achieve a fully automated sample preparation process that would be ideal for the clinical setting.
For the comparison between resins used in the IMCStips, based on the analysis, we found that the only differences in the phosphopeptides identified by PolyTi and ZrO2 resins were differences in numbers of singly versus multiply phosphorylated peptides. However, this observation could be highly dependent on variations in the sample binding environment as well as the chromatography and ionization conditions used during the LC-MS/MS analysis. These differences in types of phosphopeptides between the resins are intriguing, and further systematic investigation is needed to understand these differences and how such subtle differences could be exploited.
In summary, we established an automated workflow that combines peptide desalting and phosphopeptide enrichment within 2 h by leveraging dSPE that allows for reproducible sample preparation. The total number of phosphopeptides identified reached ~10,000 per sample without fractionation. The program was integrated onto the Hamilton STAR without additional hardware. Greater than 90% phosphopeptide specificity was achieved on PolyTi, with biochemical replicates showing similar reproducibility to that of the technical replicates, indicating that extraction efficiencies are at the detection limits of the MS. Using the established MRM assay, greater than 50% phosphopeptide recovery for all three phosphopeptides with CVs below 20% for both resin types was reported over a 3-week period. Based on these results, this automated workflow could be used to standardize the sample preparation of phosphopeptide samples.
Supplemental Material
Supplemental_Material_for_Automated_PhosphoEnrich_by_Mullis,_et_al – Supplemental material for Automating Complex, Multistep Processes on a Single Robotic Platform to Generate Reproducible Phosphoproteomic Data
Supplemental material, Supplemental_Material_for_Automated_PhosphoEnrich_by_Mullis,_et_al for Automating Complex, Multistep Processes on a Single Robotic Platform to Generate Reproducible Phosphoproteomic Data by B. Todd Mullis, Sunil Hwang, L. Andrew Lee, Anton Iliuk, Rebekah Woolsey, David Quilici and Qian Wang in SLAS Discovery
Footnotes
Acknowledgements
B.T.M. and Q.W. gratefully acknowledge helpful discussions with Drs. Michael Walla and William Cotham regarding the usage of the LTQ-Orbitrap Velos Pro system.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: L.A.L. is employed by IMCS, Inc. Q.W. is a cofounder of IMCS, Inc.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Q.W. thanks the NSF and SC EPSCoR/IDeA Program for partial financial support under NSF award no. OIA-1655740. The views, perspective, and content do not necessarily represent the official views of the SC EPSCoR/IDeA Program or those of the NSF. The Mick Hitchcock, Ph.D. Nevada Proteomics Center is supported by a grant from the National Institute of General Medical Sciences (no. GM103440).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.