
Abstract
Pharmaceutical discovery and development is a long and expensive process that, unfortunately, still results in a low success rate, with drug safety continuing to be a major impedance. Improved safety screening strategies and methods are needed to more effectively fill this critical gap. Recent advances in informatics are now making it possible to manage bigger data sets and integrate multiple sources of screening data in a manner that can potentially improve the selection of higher-quality drug candidates. Integrated screening paradigms have become the norm in Pharma, both in discovery screening and in the identification of off-target toxicity mechanisms during later-stage development. Furthermore, advances in computational methods are making in silico screens more relevant and suggest that they may represent a feasible option for augmenting the current screening paradigm. This paper outlines several fundamental methods of the current drug screening processes across Pharma and emerging techniques/technologies that promise to improve molecule selection. In addition, the authors discuss integrated screening strategies and provide examples of advanced screening paradigms.
Introduction
Drug failure rates in the pharmaceutical industry have remained high and continue to be a challenge for drug companies. Despite mean development costs of well over US$1 billion per marketed drug, approximately only 10% of drug candidates entering phase I clinical trials are eventually approved for marketing by the U.S. Food and Drug Administration (FDA).1–3 There are many reasons reported for these failures, which prominently include unpredicted drug safety issues.4,5 In 2016, for example, Hwang et al. illustrated the failure rates of 640 novel therapeutics entering clinical trials. 6 The majority of failures were due to inadequate efficacy (roughly 57%), whereas nearly 17% failed due to safety concerns. 6 Identifying such safety concerns has consistently remained a challenge for clinical candidate drugs and may be attributed to a number of factors, including difficulties in translation from animal models to humans, as well as an inability to recognize potentially susceptible human subpopulations. Also, not often accounted for are the drugs removed from development prior to human clinical trials. It is likely that many of these compounds were correctly identified as having safety issues and therefore halted from development, but it is also plausible that a portion of these drugs would have been suitable for continued development because their preclinical toxicities do not translate to humans.
Better in vitro and in silico screening may help to identify these gaps in the current testing paradigm. With more complex in vitro human models emerging, there is the potential to better predict complicated human safety issues.7–10 Producing more preclinical counterparts to these complex in vitro models will also help increase confidence in the human tissue-based systems, as differences in in vivo species sensitivity may also be evident in these models. 7 In addition, computational methods offer promise to improve the screening paradigm. It is not possible to effectively screen all potential interactions for every candidate at this time, but in silico screening is more feasible for extending off-target coverage assessments, especially for very early candidates. 11 For this reason, most pharmaceutical companies currently use a number of computational tools to predict unintended interactions. 11 However, these predictions/interactions alone are insufficient to assess the risks associated with off-targets. The therapeutic free plasma concentration of the drug must be taken into consideration while assessing the risks linked to the off-target interactions. 12 In addition, these predictions are not typically used alone for decision making, but as part of a larger weight-of-evidence assessment of potential hazards.
Polypharmacology is a well-described phenomenon that is of particular interest to pharmaceutical developers of small molecules (SMs). 13 Optimized leads generally satisfy at least three rules of the so-called “rule of 5.” 14 Because of their relatively small size, the optimized leads bind to multiple targets, including the intended pharmacological target. 15 Previous reports have suggested that typical SM drugs interact with an average of approximately 6–12 targets.11,13,15 This is particularly true at higher concentrations, which can be reflective of the exposure levels in animals during toxicology studies, or even in some patients due to variability of individual exposures or misuse. This highlights a key principle of toxicology, that all xenobiotics (including drugs) are toxic at some level. 16 Essentially all SMs will interact with additional targets at high/higher concentrations, and some will have high-affinity unintended interactions as well. A difference in affinity between the intended pharmacological target and unintended targets is an important factor determining the relevance of these lower-affinity targets, but the key comparison is to actual in vivo exposures. These exposures may be different in some human subpopulations for a variety of reasons related to genetic makeup, diet, environmental exposure to chemicals (natural or synthetic), and/or concomitant exposure to other drugs. 17 Such factors make it more difficult to definitively assess the potential safety risks of drug candidates. Whereas off-target interactions are often the main reasons behind preclinical/clinical toxicities, some drugs are intentionally designed to exploit polypharmacology for improved efficacy, particularly in select indications such as oncology, where toxicity is the desired outcome. 18 Polypharmacology has also been beneficial in repurposing existing drugs for new therapeutic indications.
According to a survey conducted by the Innovation and Quality (IQ) Consortium with members of the pharmaceutical industry on causes of drug failures, the majority of drug failures due to safety reasons are from preclinical toxicities. 19 Preclinical and clinical toxicities represent more than one-third of the overall drug attrition rate in research and development (R&D), some of which may not be relevant to humans. 19 The top reason for drug attrition has been attributed to either off-target interactions or the physicochemical properties of compounds. In addition, several factors complicate the identification and significance of SM off-target interactions in preclinical systems. These include species differences in affinity, scarcity of off-target screening assays for preclinical targets, lack of computational tools for predicting preclinical interactions (largely due to a shortage of preclinical screens), and lack of accurate tissue expression information for preclinical protein targets in some cases. Appropriately, off-target safety screens have focused initially on potential human protein targets. However, in order to better understand interspecies differences, screening against some key preclinical targets may also be necessary. This information would be helpful for putting preclinical toxicity findings, or a lack thereof, into perspective. As more in vitro preclinical protein target screens emerge, data will be created to inform computational models, and result in accurate computational model development.
Another significant focus of screening in the pharmaceutical industry is conducted to identify the pharmacological target of compounds identified in phenotypic screening. 20 Phenotypic screens identify active SMs, but by their nature do not conclusively identify the molecular interactions responsible for phenotypic effects. 20 Additional screening is required, as described in this review, to identify the molecular target(s) of action. Follow-up assessments (to phenotypic screens) for target identification may also serve to identify off-target interactions.
As previously discussed, drug-induced toxicity has been a primary cause of preclinical attrition. Orally active SMs largely exist within well-defined distributions of physicochemical space, as initially shown by the rule of 5, which includes the criteria that an octanol-water partition coefficient log P (c log P) not be greater than 5. 21 However, studies from Pfizer and Lilly agree that the risk of preclinical toxicity at an exposure level of >10 µM increases around threefold for compounds with c Log P > 3. 21 Seeking to clarify the role of physical properties in toxicity-based attrition, oral drug candidates from four large companies, AstraZeneca, GlaxoSmithKline (GSK), Lilly, and Pfizer, were examined. 21 The results demonstrated that physical properties did not influence candidates progressing to phase I versus those that failed due to preclinical toxicity, but candidates failing for clinical safety in phase I were significantly more lipophilic than those proceeding to phase II (mean c Log P = 3.1). 21 In addition, there was evidence for a link between overall physical properties and attrition in the same study as drugs with increased drug-like properties progressed from patented compounds to oral candidate drugs to oral marketed drugs more often. Several studies concur that nonselective binding (or promiscuity), which has been linked to attrition and toxicity, tends to increase with increasing lipophilicity. 21 While the evidence suggests a role for physical properties such as lipophilicity in attrition, there are many successful exceptions, suggesting that physical properties should be used more as a component of a larger weight-of-evidence argument.
Elucidation of SM Targets and Off-Targets and Mechanisms
Recent technological advances have provided unprecedented snapshots of SM-mediated mechanisms of action (MoAs). Translating these information-rich readouts, such as L1000 gene expression (GEx) and cellular thermal shift assay mass spectrometry (CETSA-MS), into robust and testable hypotheses may significantly impact SM progression through the drug discovery and development pipeline. At early stages, where it is a challenge to differentiate high-throughput screening (HTS) hits for medicinal chemistry prioritization, broader biological context may enhance the diversity and quality of the selected SMs from target- and phenotypic-based drug discovery (TDD and PDD, respectively) campaigns.22–24 Because the attrition rate for most target deconvolution methods is very high, this emphasizes the need for a cogent technology integration strategy.25,26 Ideally, the assays are not only complementary but also orthogonal to each other, providing more than an incremental value proposition. Figure 1 summarizes a process roadmap and provides a framework for applying various technology offerings. The following discussion highlights details about some of these enabling technologies ( Fig. 1 ) and emphasizes the value of data-informed decisions. The themes include (1) rule out common MoAs prior to investment in resource-intensive technologies; (2) utilize a combination of target identification strategies; (3) target identification alone does not constitute a mechanistic understanding—elucidation of the impact on relevant biology is critical; and (4) recent technological advances are facilitating target–phenotype correlations to be measured for a diversity of target classes.
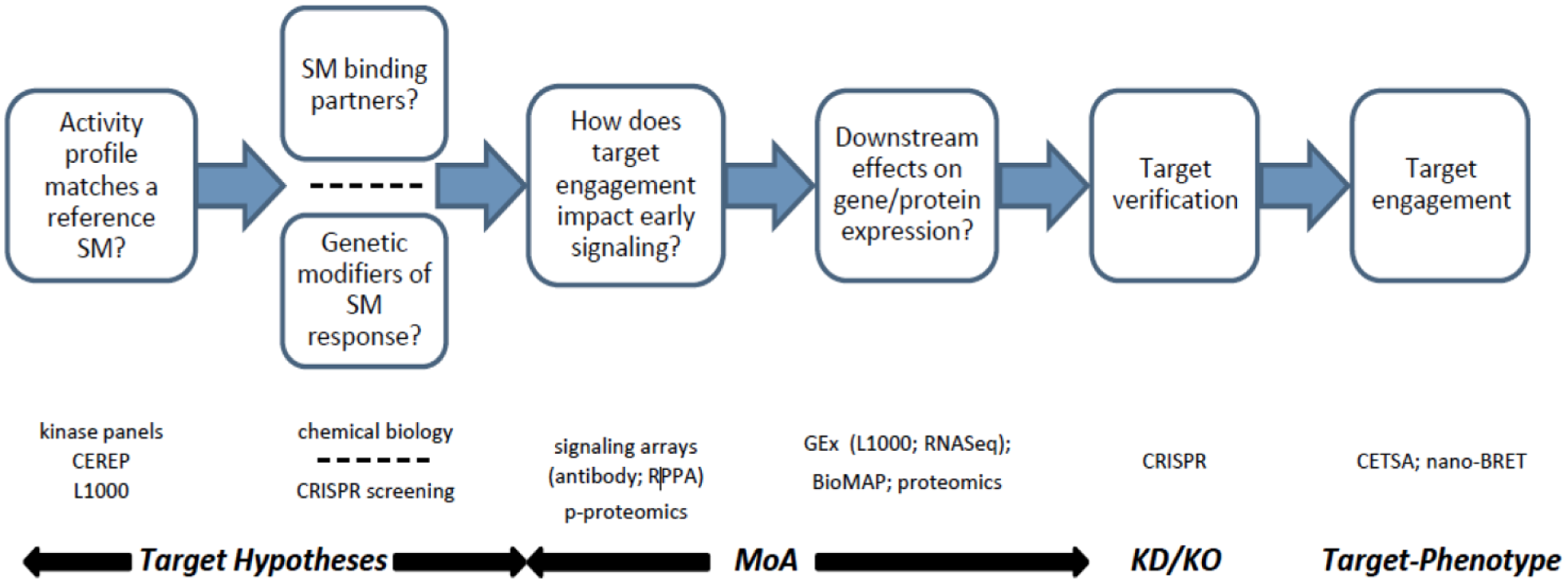
Schematic representation of a target identification strategy that supports concomitant identification of targets, off-targets, and characterization of the level of polypharmacology. Getting information about multiple targets of SM interaction at this early stage provides significant advantages for understanding and predicting later safety challenges. It can also provide a better basis for candidate selection. With the surge in phenotypic screening by Pharma in recent years, this approach gives increased understanding of drug effects during the necessary process of target/mechanism identification.
Off-Target Activity Assessment Using In Vitro Panels (Receptors, Ion Channels, and Transporters) of Ligand Binding Assays
Arguably, the most common method for predicting off-target liability is to test compounds of interest in an in vitro panel of ligand binding assays.
1
To do so, two complementary screening strategies are best employed: (1) testing against pharmacological targets that are biologically similar to the target of interest and (2) testing against a panel of off-targets known to be well associated with adverse side effects.12,27 It is also prudent to include assays for neuronal systems related to drug abuse potential (i.e., dopamine, serotonin, gamma-aminobutyric acid, opioid, cannabinoid, N-methyl-
When screening compounds of interest in various panels of in vitro binding assays, there are a number of limitations that are common to such testing. For example, HTS assays rarely measure the actual concentration of available compound in the test well. Therefore, compounds with poor solubility will often have artefactual results, appearing less potent than they actually are or even completely negative (false negatives). It is also difficult to predict off-target activity in such assays with compounds that do not bind directly to the pharmacological site of action (e.g., allosteric modulators and compounds, such as nicotinic receptor ligands, which can affect cardiovascular function indirectly via neuronal activity). 29 Prodrugs, that is, drugs that become active once they are metabolized in vivo, do not generally have much effect in these assays, and ultimately, active drug species should also be assessed. A number of assays evaluate changes in fluorescence, so test compounds that naturally fluoresce may cause interference. 30 Compounds with significant stability issues (e.g., light sensitivity) may give incorrect results when not properly protected unless care is taken to prevent degradation during shipment, formulation, storage, and testing. Some mechanisms of toxicity have not yet been defined, and others are species specific. Therefore, usage of cells from the most appropriate animal species is also an important consideration. For example, it would be a poor choice to use rodent cardiomyocytes for predicting QT interval prolongation in humans because rodents have minimal Kv11.1 (hERG) current. 31 Similarly, it should be kept in mind that most screening panel testing is conducted in cells of human origin, which may not be helpful for resolving preclinical toxicity issues. As the majority of safety failures are due to preclinical toxicities, screening against preclinical targets of the most appropriate species is an existing gap in the current screening paradigm. 19 While few preclinical protein-based screens are currently available, such screens could be used to confirm engagement in preclinical species. 32 The result could lead to a better understanding of mechanisms behind preclinical toxicities, rescuing some drugs from preclinical safety issues when these mechanisms are not relevant to humans.
When interpreting data from in vitro ligand binding panels, several aspects should be considered. Most importantly, it should be noted that these are typically assays of off-target binding affinity (and not of functional activity). Compounds may bind but have no functional activity (which our experience has shown to be somewhat rare for molecules in general) or have functional activity only at higher concentrations (which occurs quite frequently); therefore, follow-up testing of functional activity is recommended for compounds with potent binding affinity. For test compounds that have a considerable amount of plasma protein binding, in vitro potency values are usually lower than in vivo potency values because the in vitro assays generally contain little to no plasma serum. A better correlation is often obtained by using free concentration values. 33 Also confusing is when large negative values appear in the binding results: they are most commonly due to the tested compound interfering with the assay or causing allosteric modulation at the target. Lastly, compounds that bind to off-target, central nervous system (CNS)-related sites in vitro, but do not readily cross the blood-brain barrier in vivo, can still be considered for further development for non-CNS-related indications, keeping in mind the potential for issues in patients with compromised blood-brain barriers. 34
Profiling: Assay Panels
Over the last few decades, many pharmaceutical companies have invested heavily in kinase inhibitor programs. As a result, assay panels have been developed that sample kinome diversity. 35 Correlation of kinase activities that contribute to toxic liabilities, combined with access to commercially available panels that cover 450+ kinases, has enabled SMs from both kinase and nonkinase programs to be comprehensively assessed against this druggable protein class. Examples of commercial platforms include Eurofins (www.eurofinsdiscoveryservices.com/kinases), ReactionBiology (www.reactionbiology.com/), and Advanced Cellular Dynamics (discover.acdynamics.com/). In addition, these companies also offer profiling panels for many other classes of targets. However, these panels are more commonly utilized as counterscreens for targeted programs and not implemented as broad-diversity screens to characterize SM leads.
The impact of SMs on fundamental cellular processes such as cell division and electron transport chain (ETC) function during target identification or off-target screens should not be ignored. Microtubules and components of the ETC are established promiscuous binders of many classes of SMs and may be excluded using a variety of assays; however, when these activities are part of an SM’s polypharmacology profile, interpretation and action may become more challenging. 1 For example, microtubule inhibition (MTI) for an anticancer compound can overestimate tumor growth inhibition during a structure–activity relationship (SAR) campaign and lead to reduced correlations between in vitro and cellular activities. We have routinely assessed hits and leads from TDD and PDD campaigns and revealed that many chemical series from oncology and nononcology programs have in vitro MTI activity, 36 and more recently demonstrated that these activities correlated with cellular effects, including L1000 reference database matching (S. M. McLoughlin et al., unpublished data).
In the case of ETC modulators, inhibitors of complexes I and V do not exhibit acute cytotoxic effects when tested against many primary cell culture systems and often demonstrate favorable effects on anti-inflammatory endpoints.37,38 However, many inhibitors of these ETC components result in significant toxic liabilities in intact organisms and lead to lactic acidosis. 39 Unique in vitro modulation of complex I had been observed with the marketed drug metformin, including a significant decoupling of redox from proton transfer domains. 40 In spite of that, the extent of the in vivo impact on proton transfer is not without debate.41,42 The complexity of this and other related mechanisms warrants routine assessment of SMs in advanced mitochondrial cell health assays.37,38
Beyond kinases, receptors, and other target-relevant protein classes, it is impractical to assume that profiling panels will sufficiently cover the proteome. More importantly, many of these assays lack the appropriate cellular context to fully appreciate the consequences of target engagement since the systems utilized involve isolated proteins or truncated domains and mostly exclude physiologically relevant binding partners. Therefore, a more pragmatic approach to broadly assess the cellular impact of an SM is needed.
Broad-Endpoint Biological Assays with Companion Reference Databases
The inherent risk of assessing SM MoAs across a limited view of target space is potentially missing an off-target liability that would discontinue progression of a drug candidate at costly late preclinical or clinical stages. Beyond the challenge of uncovering an unknown activity for a SM of interest is the significant challenge of translating these data into meaningful decisions. Ideally, multiple readouts provide a weight-of-evidence argument for further studies related to these hypotheses. However, the depth and breadth of the profiling data must be balanced with the cost per data point and turnaround time. To address these issues, we have adopted the use of profiling technologies that measure hundreds to thousands of endpoints and have companion reference databases to permit the identification of common and targeted MoAs. AbbVie’s use of the L1000 platform for GEx and the BioMAP platform for protein endpoints has been recently described. 36 In this review, the emphasis will include application of these methods to more effectively uncover off-target liabilities. Included in this section is the use of CRISPR screening to inform on SM MoA, since we have observed this technique to directly complement data originating from L1000 and BioMAP platforms. Specifically, the use of whole-genome pooled screens when implemented with a positive selection allows for a comprehensive assessment of SM perturbations without the need to have a reference database of profiles/activities.
L1000 Platform and CMap and LINCS Databases
The Connectivity Map (CMap) tool was developed with the goal of linking diseases, genes, and SMs that modulate the relevant biology.43,44 The database initially consisted of microarray GEx profiles for a modest set of SM perturbagens across three cell lines, interfaced with pattern-matching software for data mining. This provided direct access to answering the question “Does my molecule with unknown MoA behave like a reference compound?” The value of ruling out common/suspected MoAs cannot be overstated. In other words, implementation of this approach is not specifically designed to provide robust target identification, but rather to ensure that the SM of interest is not operating by a known MoA that is inappropriate for drug development. Cellular ETC inhibition, as described in the previous section, is an example of an MoA that needs to be flagged early in SM prioritization. Another powerful application involves identifying SMs that are capable of reversing a GEx signature that is correlated with a specific cellular state, such as disease. A number of studies have successfully used this approach to identify SMs that have an anticorrelated GEx signature leading to drug repositioning45,46 and discovery of unappreciated mechanisms of tumor growth inhibition. 47 The demonstrated utility of this approach provided the impetus for expanding CMap as a community resource project. However, in order to make this a cost-effective endeavor capable of profiling large chemical libraries across a diversity of cell types, an alternative transcription profiling technology was necessary. The L1000 platform effectively solved this challenge by establishing a high-throughput, reduced-representation GEx platform. 48 Crude human lysates, derived from cells or tissue, are subjected to a multiplex ligation-mediated amplification assay in 384-well plates. A total of 978 probes representing landmark (LM) genes are directly measured for each well. Computational inference is used to correlate effects on the remaining portion of the full transcriptome. 49 Over a decade after the initial description of CMap and numerous mechanistic discoveries, the L1000 technology platform has fueled the National Institues of Health (NIH) Library of Integrated Network-Based Cellular Signatures (LINCS) program to include >1 million publicly available L1000 profiles.50,51
The L1000 platform–LINCS database continues to enable researchers to ask unprecedented questions regarding the impact of SMs on complex biology. This is exemplified by a recent study that profiled ~100 investigational and clinically approved drugs against a panel of six breast cancer cell lines. 52 The SMs were profiled in a six-point dose–response, at two time points (3 and 24 h), in triplicate, and compared with phenotypic responses using growth rate (GR) inhibition metrics. Many noteworthy observations were gleaned from this study, including a data-driven rationale for uncovering both orthogonal and nonorthogonal approaches to oncology drug synergy. It is envisaged that advances like these will help define strategies for patient selection.
In addition to dissecting the cellular response to SM treatment, L1000 data have also been used to predict SM-mediated toxicity. Kohonen et al. described a data compacting machine learning approach that combined CMap GEx data and NCI-60 cytotoxicity data for a set of 1300 SMs to create a predictive toxicogenomics space (PTGS). 53 In addition to providing information on dose-dependent cytotoxicity and predicting SM-induced pathological states from repeated dosing of rats, the PTGS tool demonstrated improved prediction of human drug-induced liver injury (DILI); however, DILI prediction continues to be a major challenge for Pharma. Robust advances in quantitative systems toxicology will continue to streamline safety testing strategies, and ideally reduce animal usage.
BioMAP Platform
The Biologically Multiplexed Assay Platform (BioMAP) consists of human primary cell-based disease models that recapitulate important cellular regulatory networks. 54 Cell models are treated with factors and stimulated to mimic the complexities of disease biology. Protein-derived endpoints, many with established clinical relevance, are measured to generate compound-specific signatures. The library of reference profiles can be used to robustly classify diverse mechanisms, such as regulation of NF-κB, and impact microtubule and mitochondrial function. 55 In the case of mitochondrial function, the MoA can be further dissected into mechanistic bins, such as those mediated by endoplasmic reticulum stress. BioMAP data provide a strong complement to GEx profiling, especially for mechanisms that fail to induce a transcriptional regulation at the time points commonly used for GEx profiling (3–6 h).
The U.S. Environmental Protection Agency’s (EPA) ToxCast program utilized BioMAP to evaluate the use of human primary cell systems to identify and classify MoAs for 641 environmental chemicals and 135 reference pharmaceuticals and failed drugs. 56 A number of correlations were uncovered that were consistent with preclinical and clinical observations. 56 For example, two PDE4 inhibitors developed for the treatment of asthma shared strong similarity to the profile of all-trans retinoic acid (ATRA). 56 In rodent studies, both PDE4 inhibitors induced fetal effects in rodent models that were consistent with the phenotype observed by the developmental toxicant ATRA.
Implementation of BioMAP profiling earlier in the drug discovery process could contribute to more informed decision making during all stages of preclinical SM selection. In our experience, SM leads from multiple PDD screens were mediating common MoAs, including impact on a kinase family of enzymes and the mitochondrial ETC. The chemistry and target deconvolution resources to support these efforts were extensive and ultimately diverted efforts away from higher-quality opportunities. We have also utilized BioMAP profiling to prioritize chemical series from the lead optimization (LO) stage for TDD programs. In a recent example, a compound from the lead series deemed “clean” in a series of profiling panels had the highest number of activities. If we had used the frequency of BioMAP activities as a metric for promiscuity, we would have misled the chemistry efforts. Since we applied a battery of complementary approaches, we were able to correlate this activity with a putative target from affinity capture mass spectrometry (AC-MS) enrichments. Thus, caution must be taken for assays that survey broad biology since “activity biases” still occur. Simply stated, SM impingement on one main target (e.g., a central transcriptional regulator) can lead to numerous downstream effects on GEx and protein expression (PEx). In cases where we failed to detect a strong database match, resources were shifted to complementary approaches.
To increase access to the technology, modified screening paradigms with reduced assay systems and fewer concentrations have been proposed. 36 The initial goal is to identify cytotoxic outputs for larger libraries of compounds using a reduced assay panel. SMs are then profiled against the full complement of 12 BioMAP assays to provide expanded mechanistic insights, and to permit fit-for-purpose assays to be implemented for medical chemistry campaigns.
SM Positive-Selection Genome-Wide Pooled CRISPR Screening
Spontaneously acquired resistance, achieved by exposing cells in culture to escalating doses of a death-inducing SM, is a common approach to uncover mechanisms that may be used by a tumor cell to develop resistance. 57 The pooled or clonal populations of resistant cells are compared with the untreated parental line, and routine analyses include GEx and/or PEx changes, as well as sequencing. When successful, this approach does not usually identify the direct SM–target interaction, but rather it identifies mechanisms that may indirectly inform on the SM MoA. However, in our experience clarity in the MoA is the exception and not the norm. Identifying the direct mediators that circumvent the SM MoA is usually resource-intensive. The advent of CRISPR technology and introduction of single gene perturbations, in combination with our existing approaches, have clarified prioritization of our follow-on resources.
CRISPR technology has significantly enhanced the technology tool set for delineation of synthetic lethality and identification of SM targets and mechanisms. 58 In cases where death-inducing compounds are used as the positive selection, multiple gRNAs for 19,000+ genes are introduced into cells and the pooled population is subjected to SM treatment at an appropriate concentration (e.g., GI90). Only the gene disruptions that confer resistance will survive this positive selection; since the majority of gene perturbations will not have a functional consequence, most of the cells die and data analyses are simplified. 59 This approach has been applied to studying the mechanism of etoposide, directly revealing the gain-of-function target, TOP2A, in addition to mediators of resistance. 59 A recent positive-selection study using rigosertib, a clinical oncology molecule with reported inhibition of PI3K and PLK, revealed that the main MoA is microtubule destabilization. 60 Positive selection beyond cell death may be achieved by enriching cells with a phenotype of interest, as demonstrated previously using pooled shRNA libraries. For example, an SM inhibitor (ISRIB) of the integrated stress response was tested for its ability to abrogate thapsigargin (Tg)-induced stress in cells transduced with a pooled shRNA library. 61 Positive selection was achieved using fluorescent reporter assays, followed by fluorescence-activated cell sorting (FACS) and deep sequencing. This study revealed the direct target of the ISRIB since it involved a gain-of-function MoA, that is, activation of eiF2B. Using a genome-wide CRISPR library, Park et al. identified a restricted set of HIV host dependency factors. 62 Genome-wide disruption of a human pediatric ALL line (T-cell lymphoblast) was followed by an HIV challenge, flow sorting, and sequencing the gRNA abundance from GFP-negative cells. In a recent example, a functional genomic screen for Hedgehog (Hh) signaling was developed by engineering an antibiotic-based selection of Hh-responsive cells in conjunction with genome-wide CRISPR-mediated gene disruption. 63 This positive-selection screen enabled the identification of factors required for ciliary signaling.
Target Enrichment
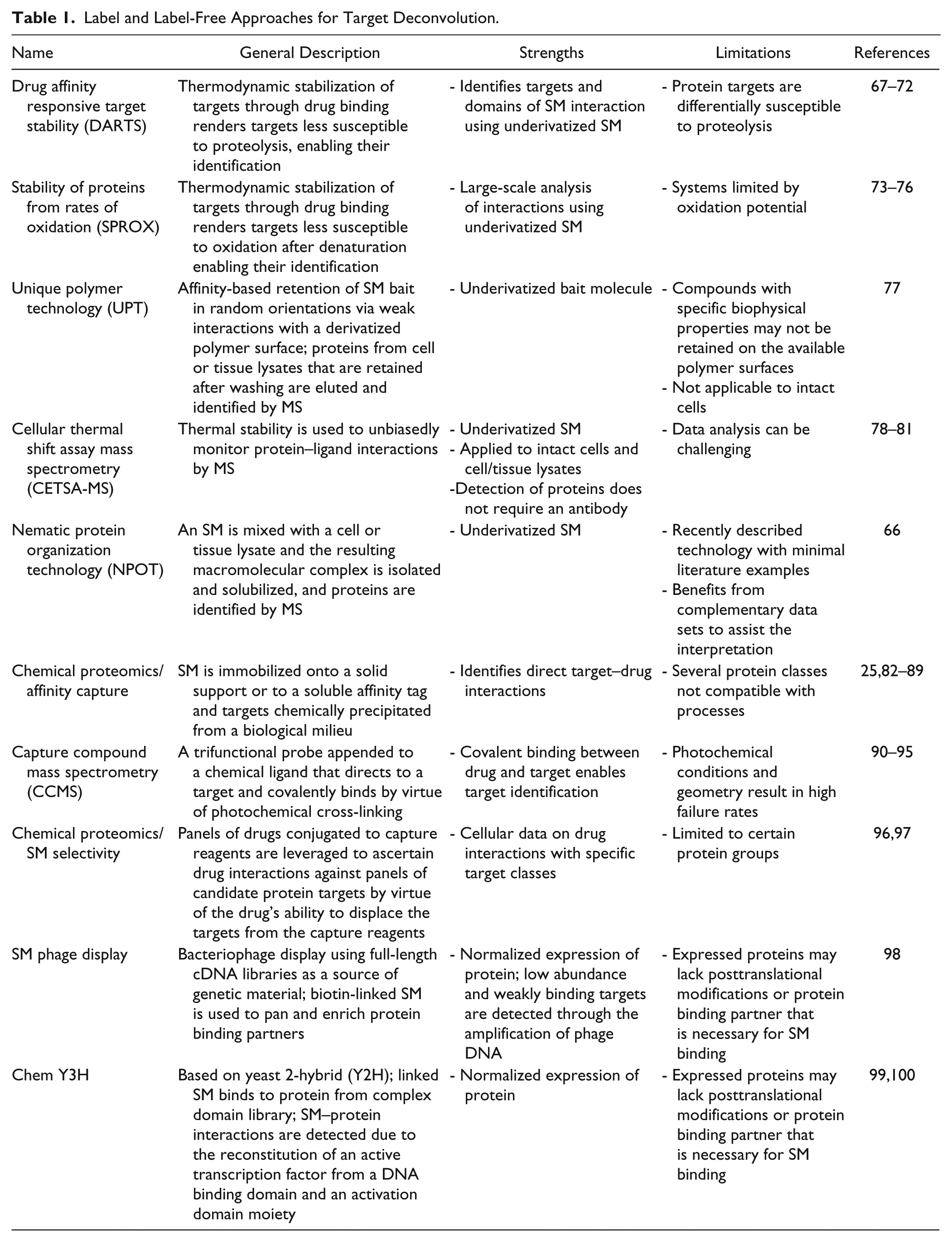
Methods that robustly identify putative binding partners for SMs of interest are highly desirable, since there is usually a clear path to hypothesis testing. Methods may be divided into two broad categories, which are primarily differentiated based on the need to chemically modify the SM with a handle for enrichment (label) and the alternative where the SM is left underivatized (label-free). 64 In the former, the SM is modified with a linker that has an affinity handle such as biotin, resin/beads, or biorthogonal functionality. Introduction of the linker is usually supported from existing medicinal chemistry efforts that determine SARs. Linkers extending off other vectors of the SM can also provide valuable information about the comprehensive target engagement profile. For example, a linker that abrogates the activity endpoint of interest may be applicable at a future time to enlighten binding partners that are relevant in other biological contexts (e.g., protein relevant to an organ-specific toxicity). Label and label-free SM–target enrichment technologies have been reviewed in detail.25,64,65 In particular, label-free technologies that only utilize the parent SM have garnered a lot of interest since they do not require synthetic chemistry resources. Table 1 is a summary of label and label-free approaches that have demonstrated utility for target deconvolution, in addition to some newer approaches for consideration. Since a single method does not exist that can consistently identify SM binding partners, and it is impractical to routinely apply a large suite of approaches, prioritizing the technologies is very challenging. Because label-free choices do not require a chemistry step, it is sensible that the straightforward offerings should be implemented. In a recent study, mebendazole was identified from the CMap database for its ability to reduce the expression of C-MYB, a transcription factor involved in acute myeloid leukemia (AML) cancers. 66 The putative target of mebendazole was investigated using the label-free approach nematic protein organization technology (NPOT), revealing data that supported a role for heat shock proteins. 66
Label and Label-Free Approaches for Target Deconvolution.
From Targets to MoA
SM target discovery studies aim to identify the protein that is responsible for either a desired or unintended response. However, this knowledge alone is not sufficient to fully appreciate the SM MoA. 26 Coupling SM–target engagement with early signaling biology may augment the interpretation of downstream gene and protein expression/regulation responses. 101 Dose and time-course studies that monitor phosphoprotein (p-protein) signal transduction, using either unbiased p-proteomics or p-protein panels such as RPPA, may reveal alternative nodes for pharmacologic modulation that are superior or do not share liabilities of the SM being studied. Furthermore, applying CRISPR-Cas9-based systems to functionalize nodes involved in cellular signaling networks 102 may ultimately aid in the prioritization of targets modulating cellular endpoints of interest. We have recently integrated p-proteomic data with positive-selection whole-genome pooled CRISPR screening outputs to identify alternative nodes for pharmacologic intervention. Hypothesis testing is currently in process.
Target Engagement and Verification
Putative targets from labeled-SM probe experiments are initially prioritized based on the ability of the enriched proteins to compete against the free, unlabeled SM. 89 In the case of label-free approaches, the profiles from active SMs may be compared with chemically similar, but biologically inactive, versions of SM. Although these data provide evidence for bona fide SM–protein interactions, orthogonal assays are usually employed. 36 In the absence of a binding or activity assay for the protein of interest, approaches such as CETSA using a Western blot output can be applied. 103 CETSA has the advantage of using the label-free parent SM, but this method requires an antibody for monitoring thermal denaturation and precipitation. We have applied this method for several TDD programs and PDD hypotheses. In the case of the latter, when enabled with a decent-quality antibody, CETSA has both confirmed MGST1 as a putative cancer target 36 and ruled out other PDD hypotheses. Although MS provides an alternative readout for CETSA that is detection reagent independent (i.e., no antibody is required), the resource requirements are more highly specialized.78–81 Current efforts are directed toward data processing, interpretation, and integration of several data sets to better understand how to utilize these data-rich outputs.
The next challenge is to link the true binding events to the phenotypic endpoint. Although target engagement may be correlated to the biological endpoint and provide a mechanistic link, modulation of the protein target is usually desired. 104 For example, creation of a knockout (KO) of the putative target mimics the pharmacologic blocking of the activity. However, this approach does not address other potential roles of target, such as a scaffolding function. Recent advances in CRISPR technology permit gene silencing/activation to test the effects of target modulation on SM function. 105
Computational Identification of Off-Target Interactions
The past few years have seen significant advancements in drug discovery technologies to identify novel compounds for a wide range of therapeutic targets.106–110 These technologies include computational methods. The computational approach typically uses two complementary paradigms: protein structure based (target centric) and ligand based (ligand centric).111–113 The former utilizes knowledge of the 3D structure of the protein as well as pocket anatomy.114,115 The latter relies exclusively on variation of biological responses with diverse ligand chemical structures, and comprises pharmacophore and quantitative SAR (QSAR) approaches. 116
Drug–receptor binding is the first event of either therapeutic or toxicity effects. Drug affinity to the target is modulated by several thermodynamic factors.117,118 One of the most critical factors is intermolecular recognition, determined by the geometric and physicochemical complementarity as well as the steric fit.119,120 The potential for intermolecular recognition is identified with a wide range of computational methods, as well as in vitro/in vivo experimental approaches.121–124
Protein Structure and Ligand-Based Approaches
Within the computational structure-based methods, SM ligand libraries are queried against 3D pockets of protein structures for spatial and chemical fitness. The fitness between the protein drug pockets and chemical structure is generally computed using automated docking tools such as Glide and AutoDock.122,125–128 These methods are extensively used in lead identification and optimization. In addition, docking methods can also be used to predict potential on- and off-targets for a given SM or a set of SMs.129–131 Briefly, in this approach, the compound of interest is computationally queried against a panel of protein structures (usually >100,000 structures) for a possible steric/electronic fit. Protein sites with a significant steric/electrostatic fit are considered possible off-targets. For example, Do et al. 132 have used the computationally docked natural products e-viniferin and meranzin against 400 manually created targets and identified PDE4 as a target for the former and COX1, COX2, and PPARγ for the latter. In another application, Muller et al. 133 used a docking approach to predict potential targets for a novel chemotype derived from combinatorial chemistry. Computational docking on 2148 selected targets identified PLA2 as the top target, and it was confirmed later by in vitro methods. Chen et al. 134 have used docking methods to predict toxicity-relevant targets for eight clinical candidates (aspirin, gentamicin, ibuprofen, indinavir, neomycin, pencilin G, 4H-tamoxifen, and vitamin C). They predicted about 83% of experimentally known side effects and toxicity-relevant targets for these compounds. However, the challenges of using this method are as follows: (1) it is computationally intensive; (2) it produces a huge number of potential targets, including false positives; (3) connecting the predicted targets to toxicity can be difficult; and (4) it is difficult to validate all the predicted targets experimentally.
Within the ligand-centric computational approach, a ligand’s chemical structure information is compared against a database of SMs with experimentally measured biological activities to identify structurally similar compounds, and thus predict the activity of the query molecule.135,136 The latter method generates a testable hypothesis very quickly. The former method is more expensive and time-consuming.
Computational Off-Target Prediction Methods
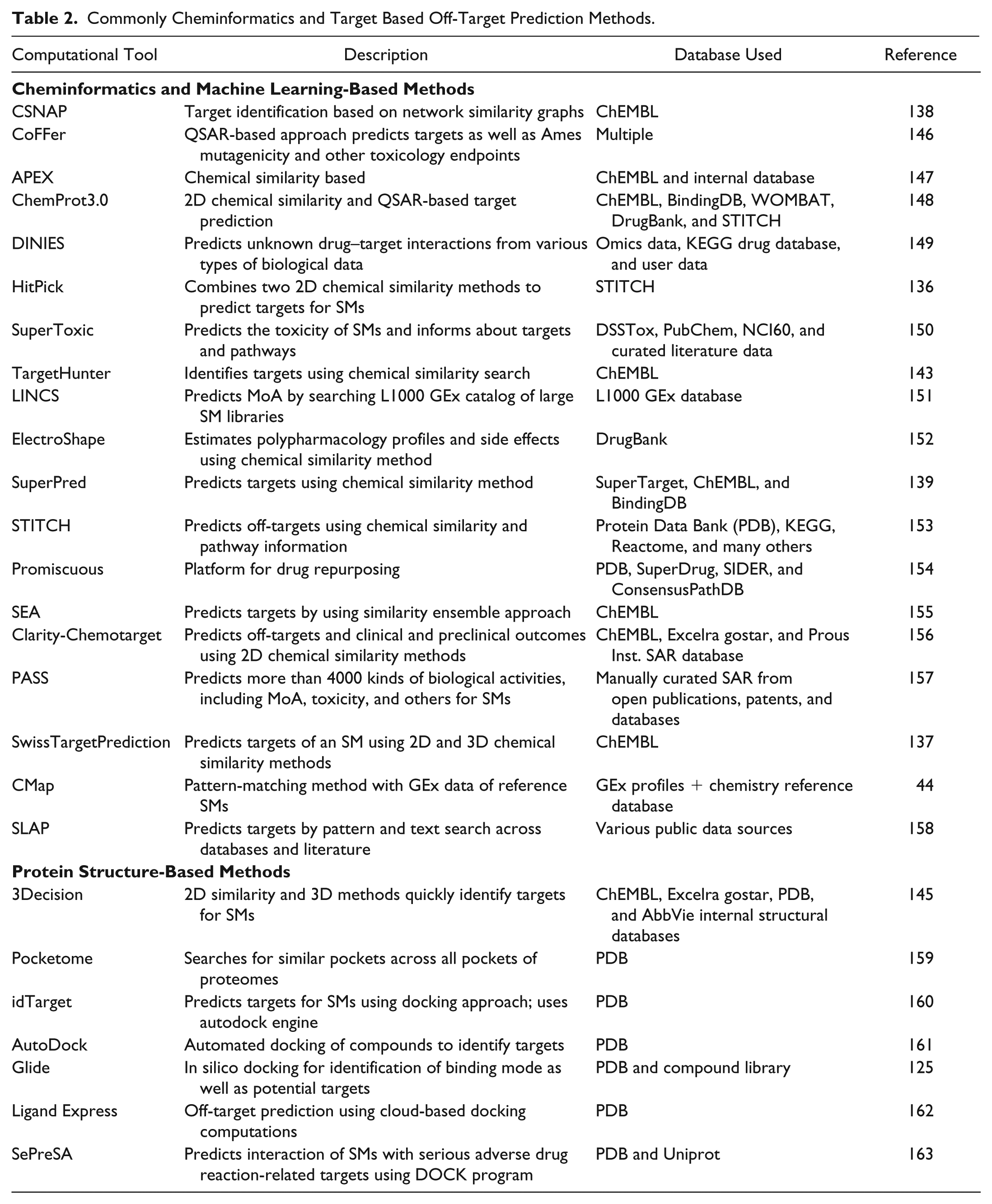
A number of computational target prediction methods, including standard tools such as SwissTargetPrediction, 137 CSNAP, 138 SuperPred, 139 DINES, 140 STITCH, 141 SEA LINCS, 142 Polypharma, 143 TarPred, 144 and other off-the-shelf modeling components (Symmetry, Chemo-targets, SEA, and CLARITY), as well as internal AbbVie tools, such as APEX, ANTELOPE, and 3Decision, are included as part of AbbVie’s novel off-target safety assessment (OTSA) approach ( Fig. 2 ). 145 Table 2 summarizes commonly used cheminformatics and target-based off-target prediction methods. Cheminformatics methods, in general, are biased by the chemical coverage of the active molecules present in the curated database. Because of this limitation, the inactive but structurally very similar compounds can display high chemical similarity with the query ligand and can produce a large number of false-positive predictions. In spite of these disadvantages, this approach rapidly predicts primary and possible secondary targets as long as the curated database contains structurally similar chemistry.
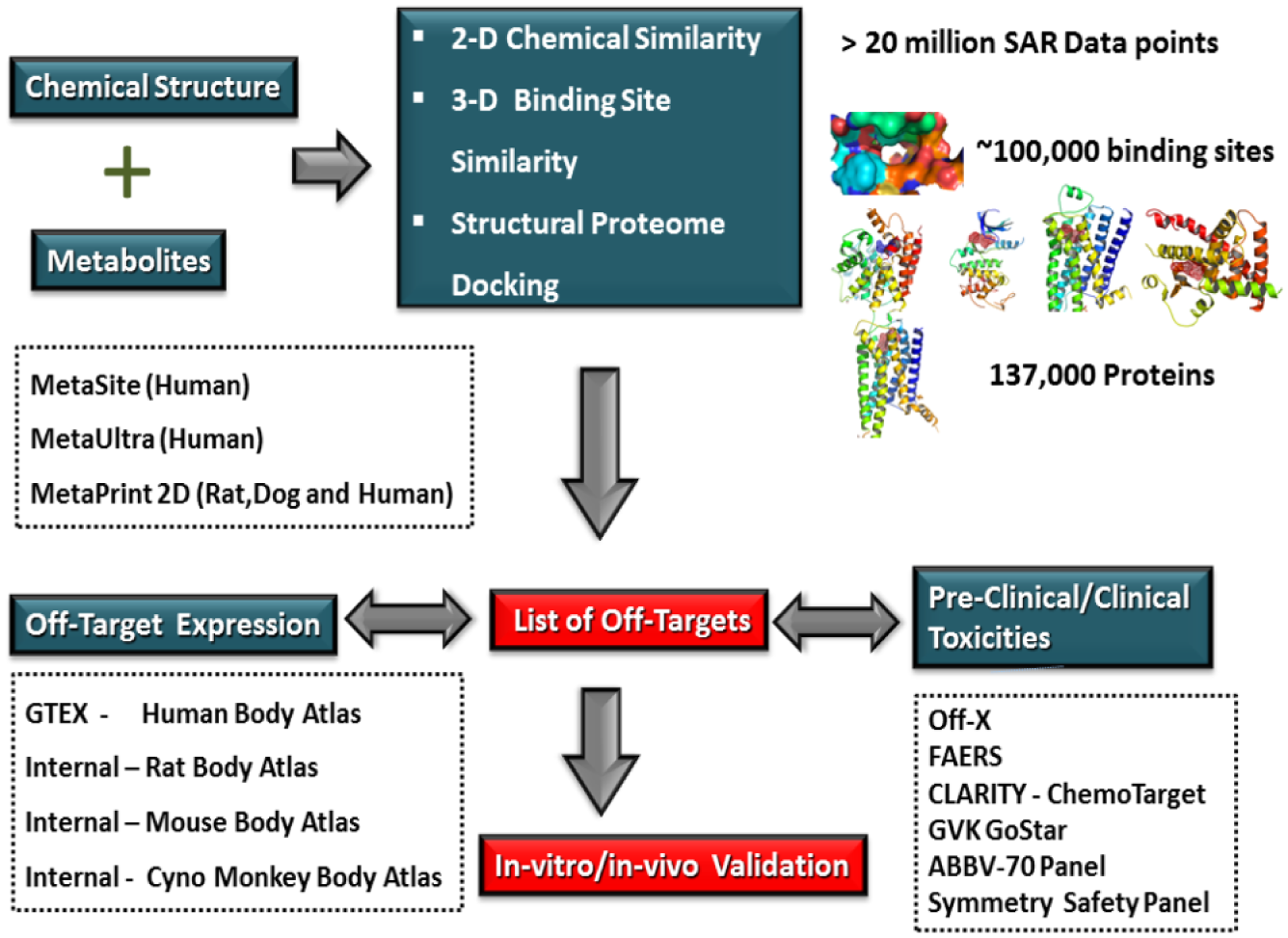
Outline of an approach to OTSA screening of SM drugs using multiple computational models. By assessing drugs and metabolites with multiple tools simultaneously, safety scientists are able to screen in silico beyond current off-target binding and kinase screening panels. The authors use a combination of 2D and 3D methods to produce a list of potential off-target interactions. These are then compared with “body atlas” GEx data from untreated species to predict potential target tissues. In addition, outcome prediction tools are used to predict the consequences of predicted interactions. Off-target interactions considered to be of potential consequence are verified and evaluated using in vitro or in vivo models.
Commonly Cheminformatics and Target Based Off-Target Prediction Methods.
On the other hand, the target-based prediction methods have proved to be successful in capturing accurate off-targets for novel chemistries (i.e., chemistry with no SAR data). They are based on the assumption that structurally similar binding sites have similar molecular functions, and thus it is likely that they bind to structurally similar compounds. Unlike cheminformatics methods, this method can be used only when either the experimental or homology model of the protein structural complex with a compound of interest (i.e., compound for which off-target prediction is needed) is available. This limits the utility of target-centric off-target prediction. One of our pursuits is the enhancement and integration of these diverse computational methods to accurately predict off-targets and associated toxicities.
Toxicogenomics: Enabling Rich Bioanalysis and Practical Considerations and Use
The field of toxicogenomics first emerged in the late 1990s with the advent of microarrays. 164 Since that time, the field has matured with the establishment of major rat databases (DrugMatrix and TG-GATES)165,166 and some enhanced understanding on the advantages and limitations of the technology. The core notion of toxicogenomics (in terms of transcriptomics) is that a toxic change will first manifest at the level of transcription, thereby either upregulating or downregulating certain mRNA, which can be queried globally. The idea of a “gene signature” or “fingerprint” is that each class of toxicity will regulate key mRNAs, and these can be compared with prototypical toxicants and drugs to empower mechanistic insights.
In the pharmaceutical industry, companies apply toxicogenomics to drug safety evaluation. Clearly, the technology can be used for a rapid evaluation of relatively straightforward endpoints, especially in rat liver, including monitoring the induction of drug-metabolizing enzymes (e.g., Cyp3a, Cyp2b, and Cyp1a), phase II enzymes, and transporters. In this manner, a xenobiotic can be identified as a Pxr, Car, or AhR agonist with relative ease using the expression of downstream genes, allowing for rapid confirmation and assessment of translatability across species.167–169
Toxicogenomics: Examples of Mechanistic Elucidation
Consequently, toxicogenomics can be most useful when applied for mechanistic insights and for predicting some toxic phenotypes, especially for those involving signal transduction and regulation of key downstream gene members. For instance, toxicogenomics has been used to elucidate mechanisms involving inflammation-associated hepatotoxicity,170–172 genotoxic versus nongenotoxic hepatic carcinogenesis,173,174 rat-specific thyroid hyperplasia, 175 immunotoxicity, 176 changes in RNA processing,177,178 and proximal convoluted tubule nephrotoxicity. 179
A key success factor for a toxicogenomics approach is the availability of comprehensive databases to compare test article-related changes with a variety of prototypical toxicants and drugs. Drugmatrix and TG-GATEs contain a wealth of data to support comparison analyses and are available for public use.165,166 In practice, differentially expressed genes (DEGs) in rats treated with a test article are compared with the entire database. A correlation analysis returns drugs or toxicants that have similarity to the database, giving clues into mechanism. Targeted follow-up investigations can then confirm mechanisms. While a toxicogenomic analysis can provide insight into toxicology studies, there are certainly limitations to the approach. For instance, existing databases are heavily focused on rat tissues and lack information on other preclinical species. Therefore, species-specific toxicities, other than rat, are not as easily interrogated by this approach. Furthermore, toxicities manifesting in organs that databases do not cover (e.g., skin, spinal cord, and testes) make it difficult to interpret data collected in these tissues. Toxicogenomics is not used to set safety margins, nor is it routinely used in traditional development enabling good laboratory practices (GLP) toxicology studies. 180 Instead, it is most often used in early, discovery-oriented studies focusing on potential mechanisms of toxicity. 180
Pathway Analyses: Insights into Toxic Changes
The most valuable interpretation of “omics” data is in the context of pathways and biological function, as opposed to individual genes or metabolites. This could also be the most difficult analysis since it can be challenging to see clear relationships for thousands of DEGs or metabolites. Pathway analysis tools and gene ontology associations are helpful for this purpose.181,182 Network-based approaches have been introduced to reduce the complexity of GEx data. 183 The TXG-MAP approach reduces complexity and helps visualize GEx data, revealing relationships that may otherwise be difficult to perceive. The authors conclude that this type of approach can supplement more traditional endpoints to arrive at enhanced study interpretation. 183 For example, Ingenuity Pathway Analysis software was used to differentiate genotoxic and nongenotoxic hepatocarcinogens and link back to pathways involved with DNA damage response and apoptosis. 184 Additionally, multiple pathway analysis tools were employed to study lipopolysaccharide-induced signaling from both in vivo and in vitro studies. 185
Toxicogenomics: In Vivo-Derived Tissues and In Vitro Applications
For toxicogenomic approaches, liver is the most investigated and well-represented tissue in public databases.165,166 The active metabolism of the liver compared with other tissues is often associated with toxic effects and makes it an excellent candidate for evaluating DEGs. Despite this, abilities to query specific cell types have great advantages. A recent investigation shows a unique molecular environment for the different zones of the liver, each adapting as needed to the microenvironment. 186 Other investigators have evaluated specific mechanisms of liver toxicity. For example, the GEx profiles of 17 compounds associated with hepatic steatosis were analyzed to identify and classify DEGs that could be used in a mechanistic signature. 187 A time-course study in the liver is often required to differentiate primary versus secondary DEGs and to identify potential pharmacological interactions.
Toxicogenomics analyses are not limited to in vivo studies. In vitro toxicogenomics can provide insight into cellular effects from xenobiotic exposure, and databases exist that hold in vitro GEx-derived profiles. 166 Several advantages can be derived from in vitro transcriptomics, including the ability to study human systems where tissue availability is limited, 178 allowing for evaluation of a full time course, studying the effects on a single cell type in isolation 178 or in the context of other cell types (co-cultures or slice models), 188 and a query of transcriptomic responses when only small quantities of a discovery candidate are available. 189 In vitro approaches also have limitations, such as low aqueous solubility of some test agents, cell systems that no longer replicate the in vivo situation, artifacts from excessive cytotoxicity, and the inability to unravel toxicities that manifest from multiorgan interactions. 170 The transcriptome of primary hepatocytes clearly diverges from the native tissue of origin ( Fig. 3 ). Some of these transcriptomic differences may be attributed to loss of cell types in culture (e.g., Kupffer cells). However, Richert et al. have shown in human hepatocytes that plating in culture for at least 24 h resulted in the most substantial transcriptomic changes. 190
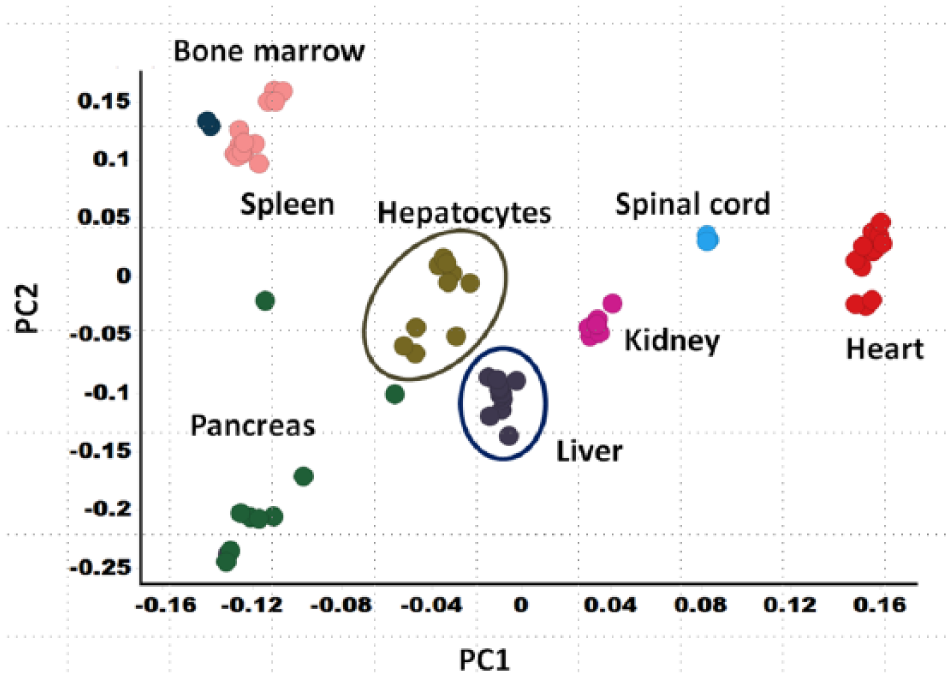
Database transcriptional profiles differ between normal Sprague-Dawley rat tissues, as expected, and result in differential clustering within principle components analysis (PCA) space. Each point represents a tissue from a single rat (either naïve or vehicle treated) as collected in an internal microarray database. Interestingly, rat hepatocytes (cultured under standard conditions for at least 24 h, either naïve or in the presence of vehicle) cluster separately from liver tissue, showing the differences in transcriptional character between in vitro and in vivo models. The complexity of the multiple tissues screened in vivo cannot currently be replicated in vitro.
As technology evolves, the ability to interrogate GEx in a small subset of cells from an in vivo thin section is becoming obtainable. For example, using laser capture microdissection, Simic et al. evaluated Notch signaling changes in the kidney microenvironment, including proximal tubules, glomeruli, and collecting ducts, after dosing rats with puromycin. 191 Notch GEx was shown to be altered in different areas of the kidney, providing insight into Notch involvement in injury. 191
Direction of the Field: Hybridization-Based Compared with Sequencing Technologies
During the last decade, in addition to advances made toward understanding the practical implications of toxicogenomics, the field has evolved to offer sequencing-based technologies.192,193 RNA-seq is a powerful application with enhanced dynamic range, greater specificity, and the ability to detect novel transcripts.193,194
Rao et al. found that RNA-seq led to enhanced information compared with microarrays when evaluating tool toxicants dosed daily in rats for a 5-day interval. 195 The correlation coefficients between transcriptional profiling data generated on the two different platforms for alpha-naphthyl isocyanate (ANIT), acetominophen (APAP), carbon tetrachloride (CCl4), and methylenedianiline (MDA) were 0.83, 0.79, 0.83, and 0.60, respectively. The RNA-seq platform also revealed GEx changes of unique transcripts, not detected by microarrays. Van Delft et al. 196 found that RNA-seq detected approximately 20% more DEGs than microarrays in HepG2 cells exposed to benzo[a]pyrene. Moreover, added insights into pathway perturbation were apparent using RNA-seq and information on DEGs at the level of splice variants. 196 Importantly, RNA-seq methods are able to assess RNA expression changes from formalin-fixed paraffin-embedded (FFPE) tissues, which could be of substantial advantage for tissues collected in preclinical toxicology studies. 197
Transcriptomics-Derived Biomarker Candidates
One clear advantage of GEx-based technologies is the unbiased identification of candidate biomarkers. 198 For instance, in one large study, candidate biomarkers were identified from mouse GEx profiles from three known genotoxic hepatocarcinogens, three nongenotoxic hepatocarcinogens, four nonhepatocarcinogens, and three compounds with equivocal results in genotoxicity testing. 199 Evaluating specific biomarker candidates, 64 genotoxic carcinogens and 69 nongenotoxic carcinogens were identified in male mice, which included functional categorization in DNA damage response, cell cycle progression, and apoptosis. 199
Some investigators have taken the approach to query a large database of GEx profiles to narrow down toxicity biomarker candidates. Kim et al. identified 18 genes associated with toxicity of the liver, kidney, and heart tissue. 200 When evaluated in cell culture, mRNA expression of neuronal regeneration-related protein was suppressed in both liver and kidney cells, while expression of cathepsin D was commonly induced in liver cells. 200 After hypothesis generation and discovery, extensive complex studies for qualification and validation of biomarker candidates are required before being adapted for regular use in toxicology assessments. 201
Toxicogenomics: Beyond the Transcriptome
As toxicogenomics has matured, it has encompassed other omics technologies for global cellular profiling of a wide variety of biomolecules.202,203 Using a parallel approach, other technologies, such as proteomics, metabolomics, and lipidomics, can be used to yield an overwhelming quantity of data for the study of a toxicological system. The application of the “correct” technologies depends on the issue at hand and toxicological phenomena under evaluation. Potentially, metabolomics can be most useful when noninvasive evaluation and time course of a potential effect is desired for the identification of SM biomarkers that can be translated between species, for rapid screening of in vivo effects against a set of either targeted or nontargeted analytes, and for mechanistic supplementation.204,205
For example, metabolomics has been applied toward mechanisms of hepatotoxicity for galactosamine and subsequent protection by glycine. 206 These data provided evidence for an increase in cellular nucleotides, thereby protecting against depletion of uridine. 206 Metabolomics has also been used to determine biomarkers after exposure to chemicals.207–210 Toxicoproteomics has been applied to understanding the toxicity of gentamicin, 211 acetaminophen, 212 and nicotine.213,214
Advanced Emerging Screening Strategies
Recent advances in in vitro models have combined multiple cell types and, in some cases, multiple tissues with microfluidics to produce the “organ-on-a-chip” platforms. Emerging models of combined tissues have followed a logical progression, with those combinations of common toxicity–disease interactions being most prominent at this stage. For example, liver- and gut-on-a-chip systems have been developed,215–217 as well as several combinations of liver and other tissues.10,218,219 Effects in the gut resulting in liver changes/toxicities have been well documented.220,221 In addition, liver combinations with other “tissues” in these models can include metabolite as well as parental drug effects, without the complication of adding rodent S9 fractions to nonhepatic models or synthesizing metabolites specifically for interrogation.10,218 Combinations of endothelial cells and tissue cell models likewise incorporate effects mediated in the endothelium, leading to wider organ damage/effects.222–225 Another common tissue combination for the organ-on-a-chip platforms includes tumor cells into liver or other common metastatic sites to assess malignancy or dormancy.226–228 Finally, combination chips with some level of immune function, in addition to other tissues, offer the promise of better identifying/characterizing human toxicities or diseases with an immune component as well.7,9,229 Even the simpler static organoid models are incorporating a number of tissue cell types in combination to provide more tissue-like interaction and function than monoculture cell systems.230,231 With the development of more complex in vitro models, an opportunity to create better computational models of human toxicity processes exists. Initial contexts for use of this technology within Pharma will likely be for the identification of mechanisms of toxicity and early screening for liabilities not easily or well tested by other in vitro methods. 7 Substantial qualification of some systems will be needed to gain adequate confidence for concrete decision making with this technology, and this may be a challenge for more expensive and lower-throughput platforms. 7 In general, throughput is low for these systems, but varies considerably across platforms.
Networks describing biologic processes such as proliferation, inflammation, cell stress, and apoptosis should also be included to afford a quantitative assessment in in vitro experiments. 232 While most current approaches have focused mainly on individual cell types, network modeling approaches representing the multiscale pharmacokinetic/pharmacodynamic processes required to describe biological responses at the tissue/organ/organism level are needed. 232 Most of the early success with this approach has been in focused specific toxicological risk assessments for therapeutic programs.
Chemical libraries can be evaluated with genomics screens to identify specific drugs, such as kinase inhibitors. For example, this approach was used to identify specific inhibitors of cyclin-dependent kinase 2 (CDK2). 233 This methodology has been termed “chemogenomics,” in which high-throughput genomics (including transcriptional profiling) of an intact biological system (such as a cell line) and chemistry disciplines are combined to identify drugs. 234 The term has also been applied to approaches including proteomics. Many leading pharmaceutical companies have established chemogenomics groups, and as a result, hundreds of novel targets and compounds have been identified. 234 Chemogenomics may offer even greater potential in the more complex organotypic in vitro models as more realistic biology is represented; however, this will add additional challenge in the interpretation as well where models are more heterogeneous. If appropriate compounds representing necessary MoAs are not included, this approach will have limited predictivity. Therefore, novel MoAs will not be easily uncovered through chemogenomics.
Key emerging in vitro platforms incorporating multiple endpoints for more accurate toxicity assessments include HTS and high-content screening (HCS). 235 HTS systems can screen individual compounds against a large number of assays to identify toxicity pathways or a large number of compounds against a single toxicity pathway assay using liquid handling robotics systems and computerized data processing. The collaborative efforts of the EPA, NIH, and FDA produced the Tox21 program to develop and demonstrate the utility of HTS to assess biological activity and identify MoAs of thousands of chemicals. 236 The ultimate goal of this program is to establish in vitro signatures of rodent and human toxicity by comparing HTS data with the historical database generated by the National Toxicology Program (NTP). 237 A testing library of ~11,000 environmental chemicals and pharmaceuticals was screened against a panel of >80 biochemical and cell-based in vitro assays. Similarly, ToxCast, an EPA HTS program, was developed for chemical safety assessments. The data from these efforts can be accessed publicly via several databases and chemical browsers. 235 Unfortunately, despite the great promise of the HTS approach, thus far results have been mixed. For example, a comprehensive evaluation of ToxCast’s in vivo predictivity showed that the in vitro assays employed were poorly predictive. 238 In addition, the authors of this study concluded that the best use of ToxCast may be for the identification of mechanisms of toxicity rather than in predicting toxicities. This conclusion is reminiscent of other technologies, such as toxicogenomics, which offered great promise as a predictive tool but has ultimately been most successful as a tool for the identification of mechanisms of toxicity. 239 HCS, in contrast, is based on modern molecular imaging technologies, which make it possible to visualize cellular and molecular processes within intact living cells. 240 HCS assesses multiple parameters simultaneously in individual cells that include fluorescent changes from molecular probes/substrates, as well as changes in morphology, migration, and cell size and number that may be associated with toxicity or adverse functional effects. 241
The systems toxicology approach is another example of integrated high-throughput large-scale data collection for screening purposes. 242 This approach combines traditional toxicology with quantitative analyses of large networks of molecular and functional (such as omics) changes at multiple levels of biological organization. Integration of multiple omics measurements across biological organization, that is, the integration of genomics, proteomics, and/or metabolomics, might be utilized as an integrated system rather than a huge collection of separate measurements. 235 Systems toxicology is based on the concept that no individual level of biological organization is sufficient alone to characterize the complexity of biological systems and changes related to disease or toxicity.243,244 However, combining multiple large and complex data sets into a coherent approach that properly evaluates each data set with the proper weight is challenging. Refining and reducing these integrated data optimally will be a significant task for the future pathway identification tools.
Summary
It is clear that there continues to be significant opportunity for improvement in mitigating pharmaceutical attrition due to adverse safety signals. 6 Several key new technologies, including those outlined in this manuscript, are lining up to help Pharma potentially improve this situation. Other important improvements that are needed include more complex predictive human screening models; corresponding preclinical models; and significant progress in analyzing large, complex, and integrated data sets from screening sets.
As the size and complexity of screening models and data sets continue to increase over time, scientists are obviously confronted with new challenges in managing and analyzing the data to get meaningful interpretations. An example of a typical screening flowchart (shown in Fig. 4 ) demonstrates this complexity. However, complexities will likely continue to increase as confidence grows in these more multifaceted in vitro models, such as organ-on-a-chip platforms. 7 As these platforms are combined with omics technologies, high-content imaging assays, drug metabolism and pharmacokinetics (DMPK), and clinical pathology readouts, the resulting data will be beyond that currently available for in vivo assessments. Instead of just snapshots of histologic changes, dynamic and complex in vitro systems may enable observation of the process of lesion formation and the pathways/processes involved with a temporal association of initiating the disease process.
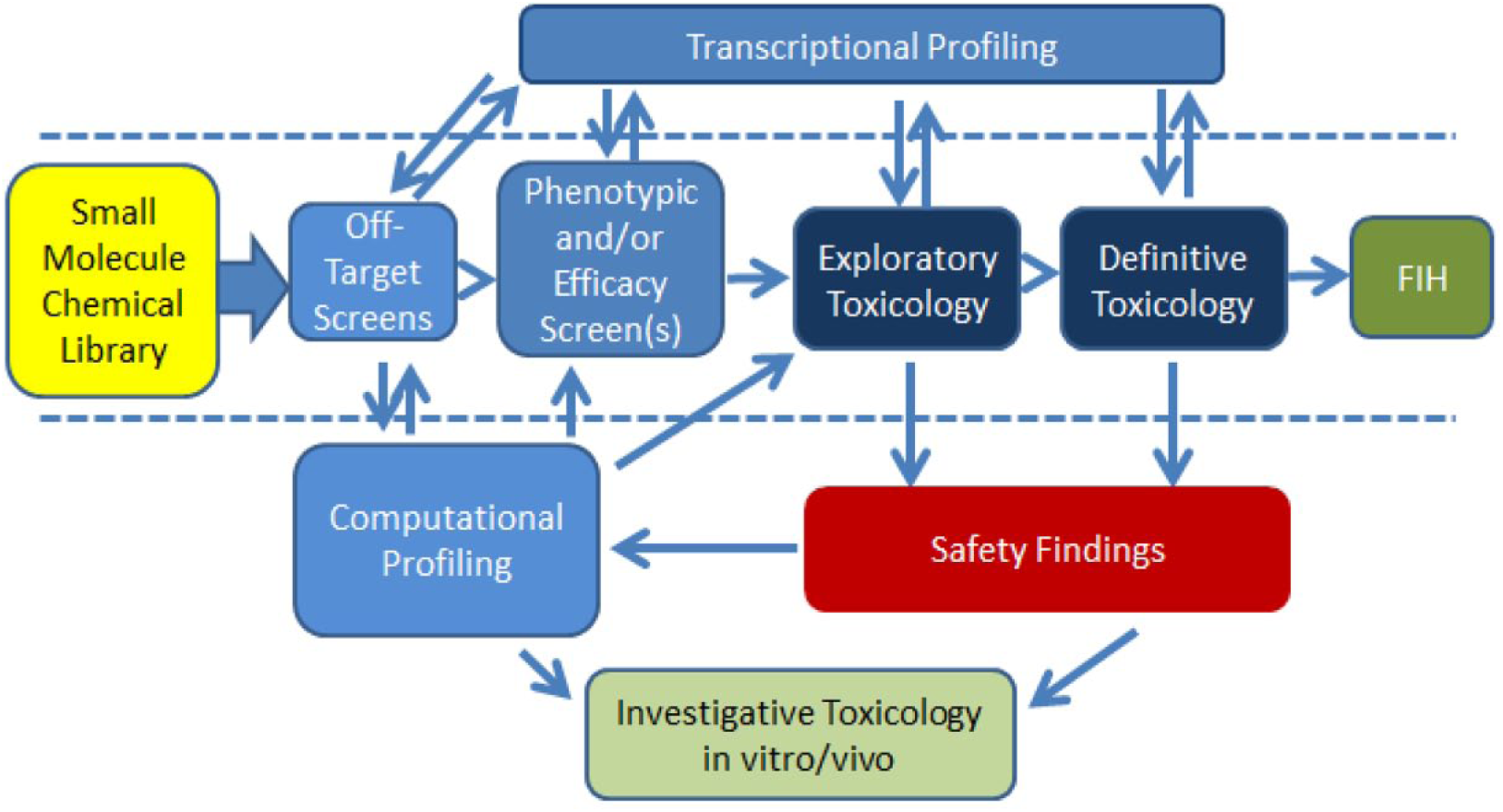
An example of what the authors consider a comprehensive drug development paradigm incorporating multistep screening and showing relationships between data that can be integrated to combine complementary data sets that provide a more complete picture of drug-related effects. A more typical minimum recommended screening paradigm is shown between the dashed lines. Combining multiple data sets with increasing complexity is a general trend in drug safety screening. Off-target screens include off-target activity panels (enzymes, ion channels, and receptors) as well as kinase panels. Phenotypic screens include broad-endpoint target deconvolution assays, CMap/L1000, BioMAP, CRISPR screens, and target engagement screens. Exploratory toxicology includes early acute and dose range-finding activities for the selection of appropriate doses in definitive GLP toxicity studies. The computation methods, combined or individually, enhance target and off-target identification activities. In addition, transcriptional profiling data (microarray or RNAseq, including other omics data) can provide important additional insight into mechanism of toxicity, particularly for effects observed in preclinical species.
The concept of adverse outcome pathways (AOPs) was proposed by the Organisation for Economic Co-operation and Development (OECD) as a way to clearly define known mechanisms of toxicity. 245 AOPs start with an initiating molecular event and result in a subsequent chain of resulting events at various stages of organization, culminating with an adverse effect to the organism. 245 By defining AOPs in this way, they are amenable to computational modeling, which is an ultimate goal of the process. 246 From a regulatory perspective, AOPs outline well-established mechanisms for reference and application to human risk assessment. 247 Criticisms of this approach include the lack of inclusion of metabolism and distribution, the unidirectional nature of AOPs, and the lack of a durational component, which can be critical in the formation of certain toxic conditions. 248
The need to integrate and handle bigger and more complex data systems appears to be part of the future of safety screening and mechanism identification investigations. 249 This is a natural scientific evolution based on the increasing complexity of models and data types collected, particularly for the investigation of toxicity mechanisms. Better tools will be needed to integrate these multiple complex data sets and analyze them. The ongoing movement from hybridization-based transcriptional profiling to next-generation sequencing-based profiling will add the complexity of splice variants, microRNA (miRNA), and long noncoding RNA (lncRNA) data to transcriptional information. 250 Significant information (such as miRNA) from sequence-based transcriptional profiling efforts is not currently being fully leveraged within typical mRNA assessments, and as a better understanding of other noncoding RNAs, such as lncRNA, develops, tools will need to advance to support their use/application as well. The collection of this supplemental data in conjunction with other variables will likely boost understanding of the roles of these additional transcriptional components in toxicology processes as well. Fortunately for screening, simplifications of transcriptional profiling methods and data, such as L1000, may partially balance the trend toward increased size and complexity. 51
An existing gap in safety assessment remains the availability of adequate target screening for preclinical species. Because preclinical toxicities are responsible for the majority of safety failures for new drug candidates, a strategy for understanding the species translatability is key to developing any of these candidates that may be suitable for human use. 19 The development of improved human models is rightly the first step in that process, but it is difficult to have confidence in novel human in vitro models at a level that would justify dosing or continued dosing in humans in the presence of significant preclinical toxicities at an unacceptable safety margin. The ability to differentiate preclinical species from humans on the basis of target/off-target interactions helps to add proper perspective to likely translatability. Differences in the expression pattern or binding affinities between species for identified protein targets may provide additional opportunity to estimate/understand the human translatability of associated toxicities. Models incorporating binding affinity data or toxicity endpoints from in vitro models can be combined with drug concentration data and in vivo exposure information to generate reliable mathematical models to predict species and particular human toxicities. In this review article, the authors have described typical safety screening options and other components for more thorough paradigms for new drug compounds. In addition, we have discussed many emerging technologies, including in vitro platforms, computational approaches, transcriptional technologies, and several approaches combining multiple technologies. For investigating mechanisms of toxicity for human risk assessments, the newer and more complex models, methods, and multifaceted data sets may provide great impact. Models such as the microphysiological systems will likely fill needed screening gaps and allow functional assessment of the glomerulus, bile ducts, endothelium, and nerve. These tissues have not been easily modeled in vitro and represent a great opportunity for improved safety assessment. As experience, confidence, and improvement/modification with these technologies advance, some/many will likely be adapted to higher-throughput formats and become key parts of tomorrow’s standard screening process.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of AbbVie. The support for this research was provided by AbbVie. AbbVie participated in the review and approval of the publication.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.